海归学者发起的公益学术平台
分享信息,整合资源
交流学术,偶尔风月

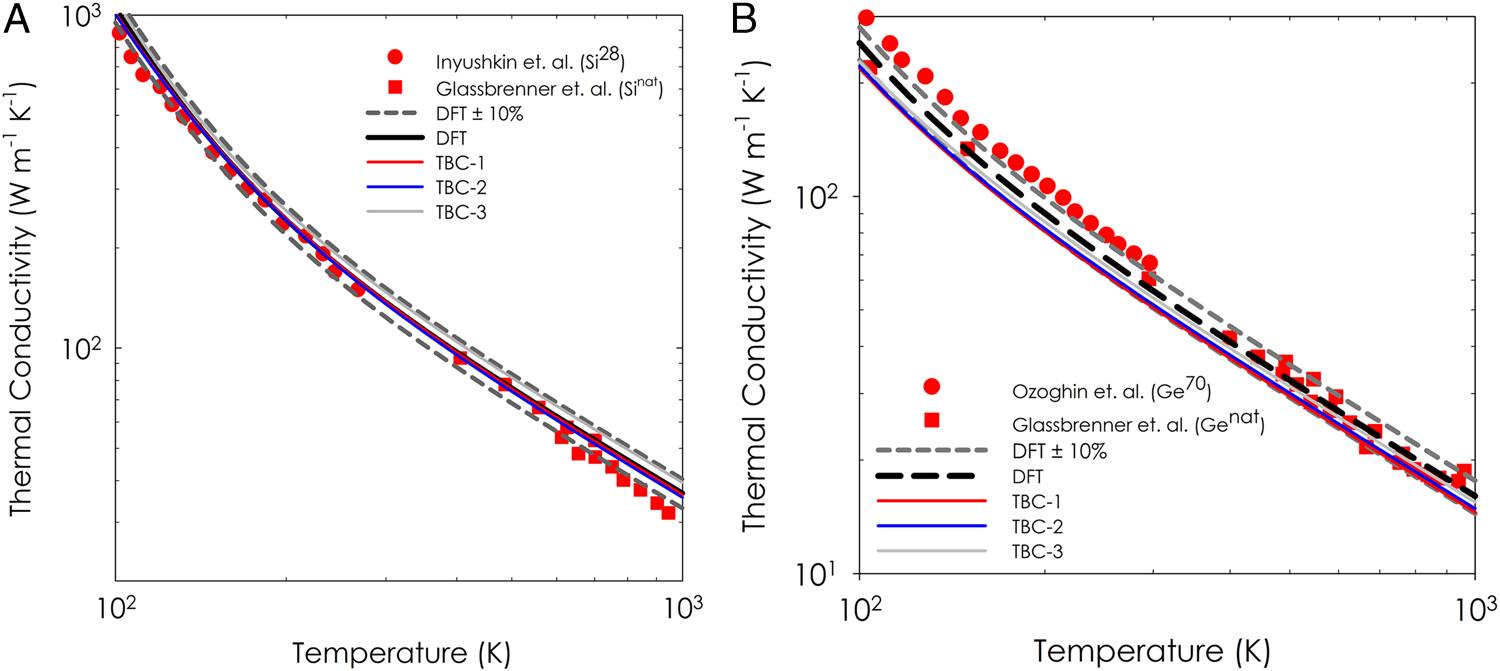
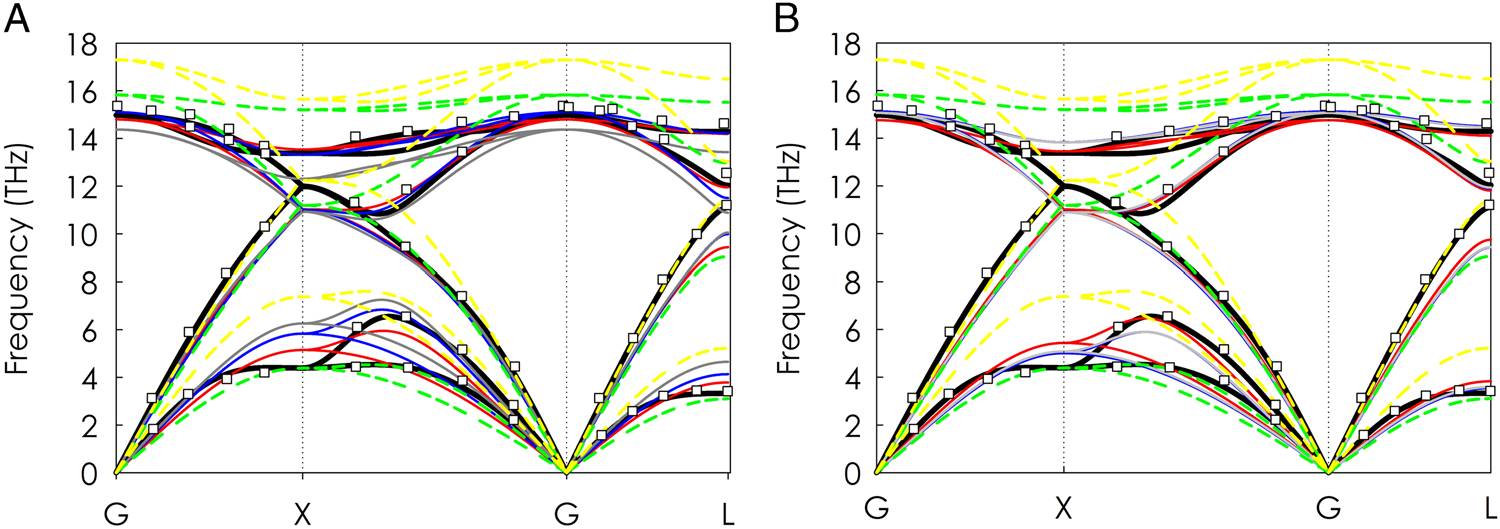
分子动力学模拟已广泛用于声子研究并取得了较好的进展,但通常因缺乏针对具体体系的准确的经验性原子间相互作用势,而难以与实验数据进行定量比较,因而无法计算和理解诸如热导率等材料性能,成为当前原子水平模拟应用的重要障碍。美国佐治亚理工学院的Asegun Henry教授领导的国际研究小组,基于遗传算法和第一原理的结果为输入,优化了经验性原子间相互作用势,使经典分子动力学模拟可用来更精确地研究声子。该方法在半导体硅和锗上得到了证明,并有望延伸应用到合金和无序等系统中。该文近期发表于npj
Computational Materials 3
:27 (2017); doi:10.1038/s41524-017-0026-y;
标题与摘要如下,论文PDF文末点击
阅读原文
可以获取。
Empirical interatomic potentials optimized for phonon properties
(针对声子性能研究优化的经验性原子间相互作用势)
Andrew Rohskopf, Hamid R. Seyf, Kiarash Gordiz, Terumasa Tadano & Asegun Henry
Molecular dynamics simulations have been extensively used to study phonons and gain insight, but direct comparisons to experimental data are often difficult, due to a lack of accurate empirical interatomic potentials for different systems. As a result, this issue has become a major barrier to realizing the promise associated with advanced atomistic-level modeling techniques. Here, we present a general method for specifically optimizing empirical interatomic potentials from ab initio inputs for the study of phonon transport properties, thereby resulting in phonon optimized potentials. The method uses a genetic algorithm to directly fit the empirical parameters of the potential to the key properties that determine whether or not the atomic level dynamics and most notably the phonon transport are described properly.
本文系网易新闻·网易号“各有态度”特色内容
欢迎广大学者供稿,报道最新研究成果
 投稿、授权、合作事宜请联系
投稿、授权、合作事宜请联系
[email protected] 或微信ID: scholarset
回复“
目录
”或“
分
类
”,浏览知社更多精华。长按二维码识别,可以关注/进入公众号进行回复。




