1. 在拟南芥中过表达TET1使基因组低甲基化
首先, 本文作者构建了在CaMV35S启动子控制下表达人类TET1蛋白催化结构域(hTET1cd,残基位置1418-2136)的转基因拟南芥,并对两株独立衍生的转基因拟南芥(35S:TET1-1和35S:TET1-2)进行了全基因组测序(whole-genome bisulfite sequencing, WGBS)。WGBS结果显示:1)相比于CHG和CHH甲基化,hTET1cd表达主要对CG甲基化(mCG)产生影响:与野生型(mCG平均水平18.2%)和met1突变(mCG平均水平0.5%)个体相比, hTET1cd的表达导致了基因组中等水平的CG甲基化(mCG平均水平分别降低至35S:TET1-1的8.9%和35S:TET1-2的6.9%, 图1a);2)在基因和转座子处都观察到mCG的强烈减少和mCHG / mCHH的轻度减少(图1c-1i);3)低甲基化现象在基因中比在转座子中更显著(图1e-1f, 补充数据1-2)。
2. TET1介导的DNA去甲基化被证实可模拟met1突变体
为评估hTET1cd表达对全基因组的影响,作者随后对差异甲基化区域(differentially methylated regions, DMR)进行了系统分析, 共鉴定出56,283个CG DMRs,其中38.7%位于基因间序列(Intergenic)中, 53.7%与基因重叠,7.6%位于启动子区域(位于基因上游≤1kb)。 与在met1突变体中观察到的结果类似, hTET1cd表达的主要作用是诱导CG低甲基化。在35S:TET1-1和35S:TET1-2中, 分别有12,641和20,601个DMRs上mCG降低超过50%;但整体上hTET1cd过表达引起的mCG降低程度仍低于met1突变体。
过往对met1甲基化组的研究曾揭示了一个CG低甲基化区域亚群中mCHG/mCHH的缺失17。在这些基因位点上,DNA甲基化能够稳定地被消除。这些位点是epimutagenesis的理想目标,因为1)与单独的mCG相比,同时具有三种类型的甲基化位点被证明更频繁地与基因的转录抑制相关;2)这些位点上低甲基化的长期稳定性能够促进可遗传的转录改变。为了确定如果mCG被耗尽,上述区域中有多少位点易丢失非CG甲基化,作者在野生型和TET1转基因个体中创建了mCHG和mCHH水平的频率分布(图1h,i)。在35S:TET1-1和35S:TET1-2中,分别有2341和3447个区域失去了超过10%的mCHG,其中有约70%的位点与met1变异体中mCHG的下降位点重合。同理,分别有2475和3379个区域失去了超过5%的mCHH,其中有超过75%的位点与met1变异体中mCHH的下降位点重合。值得注意的是,与模型植物拟南芥相比,水稻作物基因组中含有更多mCG, mCHG和mCHH介导的沉默靶点,因而hTET1cd的异位表达是创造epiRILs的一种可行方法。
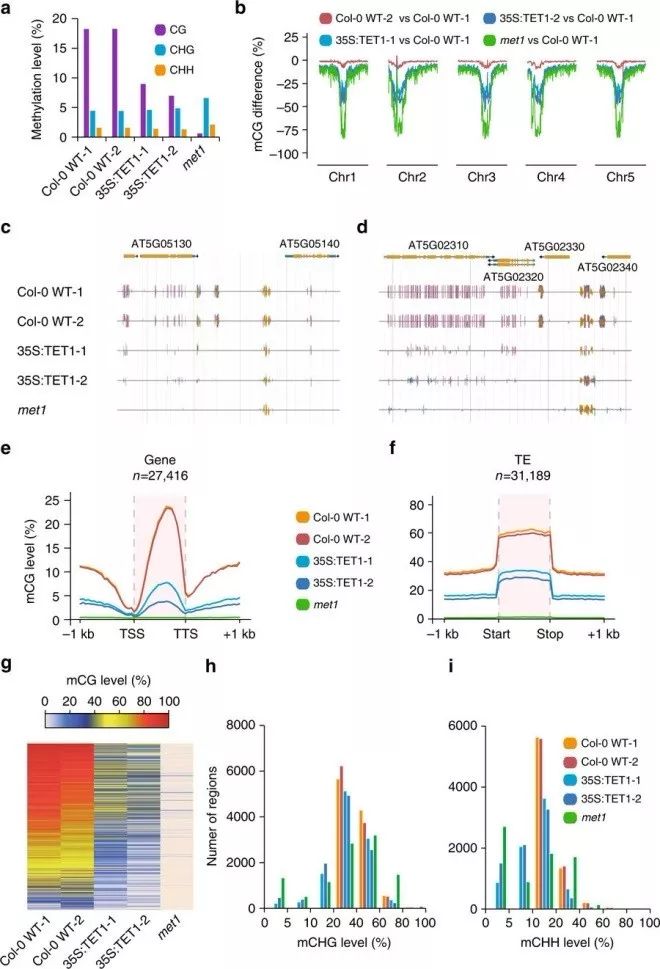
图1. 在拟南芥中过表达hTET1cd介导的CG, CHG和CHH去甲基化
3. TET1介导的CHG甲基化改变
由于histone 3 lysine 9(H3K9)去甲基酶
IBM1
第七内含子上的甲基化缺失,met1基因的突变也导致了基因体CHG位点的高甲基化。IBM1第七内含子上的甲基化缺失导致了
IBM1
的选择性剪接, 最终产生非功能性基因产物(IBM1-S),进而导致了H3K9二甲基化(H3K9me2)在整个基因组中的异位积累。与在met1变异体中观察到的结果一致,IBM1的第七内含子同样在35S:TET1-1和35S:TET1-2中被低甲基化(图2a)。 随后,作者将35S:TET1-2分别繁殖两代和三代得到35S:TET1-2
T5
和35S:TET1-2
T6
。RT-qPCR结果证实了IBM1-S转录物丰度在35S:TET1-2T6中的增加,进而引发了gene body methylated(gbM)位点上的CHG超甲基化(图2b)。为了测试功能性IBM1对H3K9me2降低的影响,作者对35S:TET1-2
T6
中的H3K9me2进行了染色质免疫沉淀(ChIP),结果显示gbM位点上H3K9me2的细微增加对应了CHG的超甲基化(图2d)。
为了进一步表征CHG的差异性甲基化,作者将在35S:TET1-2T5鉴定出的CHG DMRs根据其在野生型个体中的DNA甲基化状态分为三组。 在鉴定出的9917例CHG DMRs中, 1460个DMR位于gbM位点, 584个DMR位于非甲基化区域, 6940个DMR位于RNA-directed DNA methylated(RdDM)区域(图2e-g)。 有趣的是,在gbM位点上的35S:TET1-2T5 CHG DMRs有1409(96.5%)例表现为CHG超甲基化;而在RdDM区域中的CHG DMRs有2680(38.6%)例表现为CHG甲基化缺失,仅有825(11.9%)例表现为获得CHG甲基化。另外,有503个(86.1%)在野生型中未被甲基化的位点在35S:TET1中获得了mCHG, mCG和mCHH的甲基化。上述结果表明,epimutagenesis技术可同时诱导DNA甲基化在基因组水平上的增加和减少。
为了表征hTET1cd介导的甲基化组变化对基因表达的影响,作者分别对野生型和TET1转基因组的叶组织进行了RNA测序(RNA-seq)。 与野生型相比,在35S:TET1-1和35S:TET1-2中分别鉴定出629和736个上调基因, 其中176和260个基因与CG DMRs重叠。另一方面,在35S:TET1-1和35S:TET1-2中分别鉴定出1277和1428个下调基因,其中268和324个与CG DMRs重叠。 值得注意的是,在35S:TET1-1和35S:TET1-2中观测到的转录组变化与met1和ibm1变异体相比存在较高程度的重叠(图2h,i)。综上,在拟南芥中表达hTET1cd可以有效调控基因表达, 进而获得隐藏的等位变异来源的转基因植物株。

图2. 在转基因拟南芥35S:TET1中CHG甲基化水平的整体波动










