生信草堂
将会与更多的优秀微信公众号合作,把最优秀的微信推文呈现给大家,希望可以帮助读者更多的了解生信技术,培养和提高读者的生信分析能力!
号外,号外,号外
你想和生信分析大神做好朋友么?
你想认识更多爱好生信分析的小伙伴么?
你想让自己的生信分析走上快车道么?
那就赶快加入我们的生信交流微信群吧!
正确加入我们的模式是:
添加我们的微信Edison686868或者mly-1800为好友
标注“加入生信草堂交流群”
在群里请大家注明自己本名,单位,研究领域
便于小编管理
ActionFree 关注常见复杂疾病的遗传学研究,分享科研思路,传播生物信息分析方法,云集各路学习资料,带你了解大牛实验室,助你的科研道路妙趣横生~戳这里ActionFree公众号原文,请多关注哦~
前一段时间Jonahtan Pritchard在Cell上的文章——复杂性状扩展视图:从基因多效性到泛基因模型(An Expanded View of Complex Traits: From Polygenic to Omnigenic,放过我的翻译吧)引起了很大的轰动。 小编在反复读了原文,查阅了相关的参考文献后,以作者Yang I Li在BIRS 2017的报告为纲要,跟大家分享下自己的理解。
注:BIRS (Bnaff Internation Research Station for mathematical innovation and discovery) 【详见:2017年报告 http://www.birs.ca/events/2017】
温馨提示:请点击图片看更清晰的版本哦~

作者包括Stanford大学的Jonathan Pritchard教授,他的研究生Evan Boyle和博后Yang I Li。其中Jonathan Pritchard是小编特别喜欢的科学家,他是斯坦福大学生物和遗传学部的教授,同时是Howard Hughes Medical Institute的研究者,著名的群体遗传学家,开发了用于研究人群结构的STRUCTURE算法,以人类进化为主要研究兴趣,尤其是在理解遗传变异与人类性状之间的关系方面。此外,可变剪切的工具LeafCutter也出自Pritchard的课题组博后Yang I Li的工作。【忍不住给个特写☞↓ Jonathan Pritchard教授 】

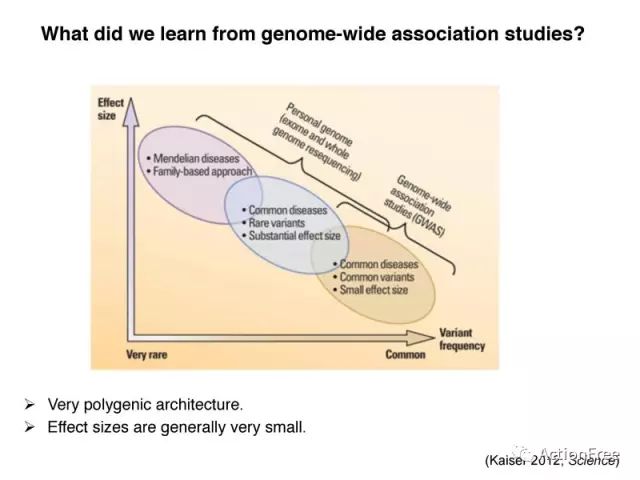
2012年发表在Science上的研究,根据effect size和 变异频率之间的关系,给出了图中的模型,即孟德尔疾病多由于具有较大效应的罕见变异导致,此类研究多采用家系研究的方法鉴定致病基因;而对于常见疾病,主要是效应较小的常见变异和一些具有较大作用的罕见变异作用产生的,即“常见疾病-常见变异”模型。自2005年第一个GWAS研究以来,得益于GWAS研究的进展和外显子测序数据的增加,在理解复杂性状的遗传基础方面让是我们惊讶的是,GWAS得到的信号,即使是那些最重要的信号,也只有非常小的effect size。

GWAS的显著的信号,只能解释很小比例的遗传度,这就是2008年发表在Nature上的 missing heritabtlity的问题。

但是如果把所有的常见的SNP都考虑进来,却可以解释很大比例的遗传度的问题,这说明目前得到的显著的位点还远远不够,可能需要更大的样本量才能得到更多更全面的显著性位点。
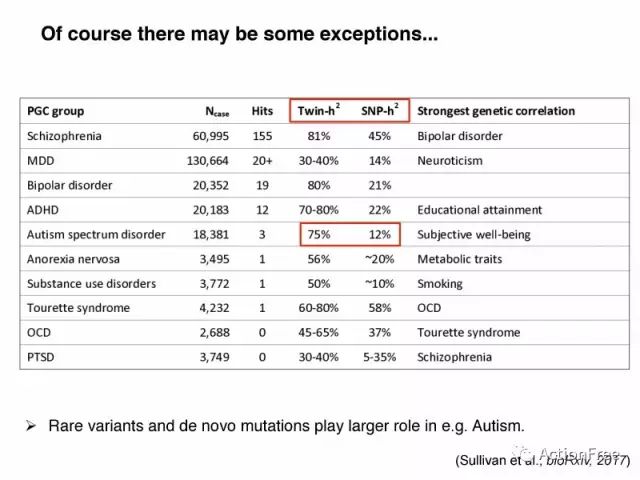
当然也有例外,比如自闭症谱系障碍(ASD)的研究,常见变异只能解释很小的比例。这说明具有较大效应的罕见变异和新发突变对于复杂性状也有重要的作用,尤其是像Autism和SCZ这种社会适应性受到较大影响很难生育的疾病。
图表解读:该图表来自Sullivan共享于bioRxiv上的文章Psychiatric Genomics: An Update and an Agenda. 用于展示目前为止PGC的9个项目组在GWAS研究中达到的样本量和主要的发现。Ncase=是case的个体数. Hits=达到全基因组显著性的独立的位点. Twin-h2 =利用同卵双生的双胞胎的共患病率估计的遗传度. SNP-h2 =利用GWAS研究结果估计的遗传度。高亮:可以看到 ASD的Twin-h2为75%,而利用GWAS的研究得到的遗传度只有12%。说明常见变异在ASD的发生发展中贡献的遗传度较少,用于支持观点:罕见的变异和新发突变在一些复杂形状中发挥重要作用。
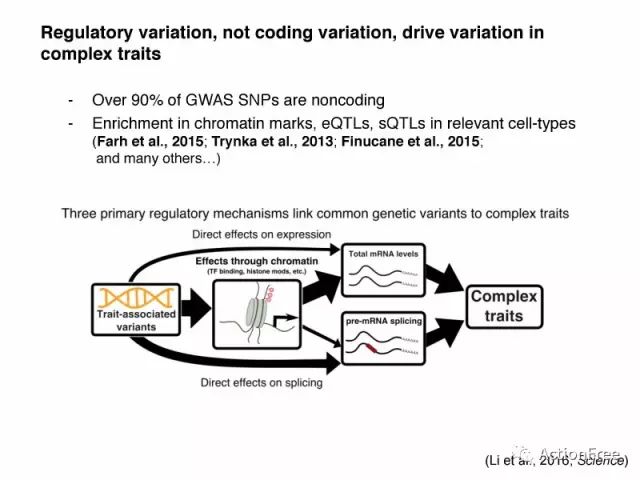
跟孟德尔疾病多影响编码蛋白质的基因不同,GWAS得到的SNP有90%是位于非编码区的,而且这些SNP在一些特定细胞类型的染色质的标记、eQTL、sQTL信号中富集。2016年Science的一篇文章给出了常见变异作用与复杂性状的三种可能的机制:
SNP直接作用与基因表达,进而影响性状;
SNP通过影响染色质的结构,染色质与TF的结合位点等,简介影响基因的表达量或基因的剪切本而影响性状;
SNP直接影响可变剪切,从而影响性状。
问题:那这些结果对于我们理解疾病提供了什么线索呢?
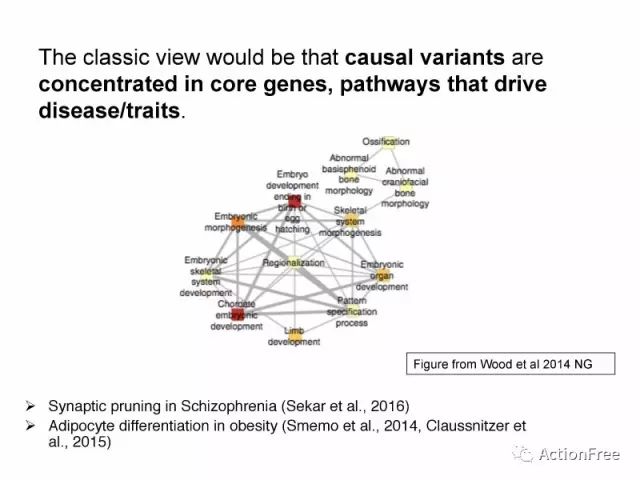
经典观念认为,致病变异(causal variants)主要集中在关键基因和通路中,如精神分裂症的研究中的突触修剪相关的通路,肥胖的研究中的脂肪分化相关的通路。
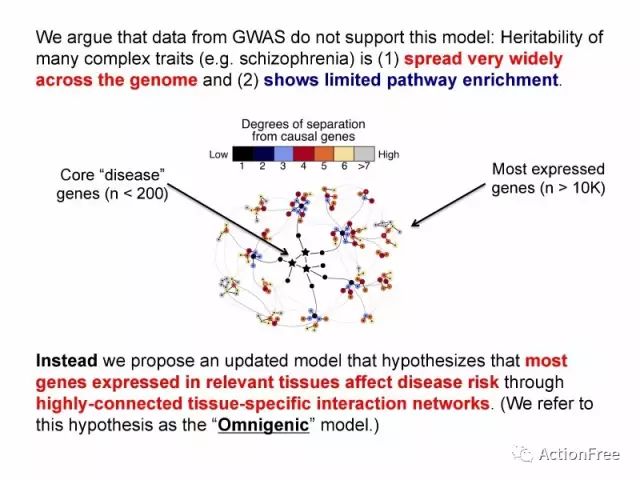
但是研究者发现GWAS的结果并不支持这样的模型(不支持如上数据的模型),以精神分裂症为例,可以解释遗传度的SNP遍布整个基因组,同时通路的富集结果非常有限。
因此研究者提出一个更新的模型,此模型的假设是:在疾病相关的组织中表达的大部分基因都会影响疾病的发病风险,这种效应通过一种高度连接的,组织特异性的网络来实现。这就是所谓的泛基因模型。关键词:表达的基因,特定组织,高度连接&组织特异性的网络 也就是说,泛基因模型的基本假设是:疾病风险是由于在特定组织表达的基因通过组织内的高度连接的网络导致的。
图片解读:degrees of separation from causal genes (degree)的含义:距离网络的核心基因的程度,degrees越小表示在网络中越靠近中心的位置,也就是中心性越高。
此图展示了在网络模型中,那些核心的疾病相关基因(小于200个),而大部分基因在网络的外周的这些基因数目庞大(大于1万)。
而在小世界网络中,每个外围的基因都可以通过很少的相互作用而对核心的基因产生影响,从而可以将这种影响传递,可以说是牵一发而动全身。
现象1:对于很多性状来说,致病区域几乎在整个基因组上,也就是并没有特定的区域贡献了特别多的效应。如下三张slides的图片,分别举例三个例子:
1.以精神分裂症的研究为例,解释的遗传度的多少跟涉及的染色体的区域大小呈线性关系;
2.扩大到30多个复杂性状的研究,不同染色体上解释的遗传度的多少似乎依旧与染色体的长度呈正相关;
3.以身高的研究进行深入的分析,GIANT项目和HRS项目的数据结合,发现若要解释身高在人群中的变异,同样需要数目庞大的SNP。
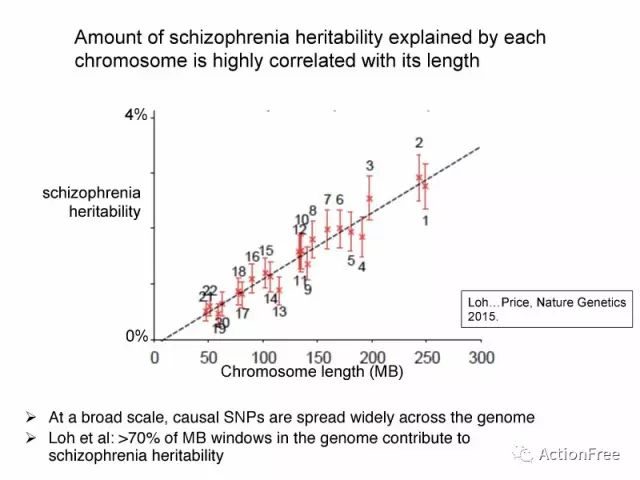
以精神分裂的研究为例,研究者发现,每条染色体解释的遗传度得多少跟染色体的长度高度相关。也就是说这些贡献遗传度的所谓致病的SNP并没有集中在某一个区段,而是在整个基因组上都有,而且以MB为一个观察区,Loh等得文章指出:基因组上有大于70%的MB范围的区域贡献了精神分裂症的遗传度。
图表解读:此图来自loh等发表在Nature genetics的文章,表示了染色体上的长度跟贡献遗传度的线性关系,x轴为染色体的观察窗口的长度,y轴为:SNP-heritability explained per chromosome,
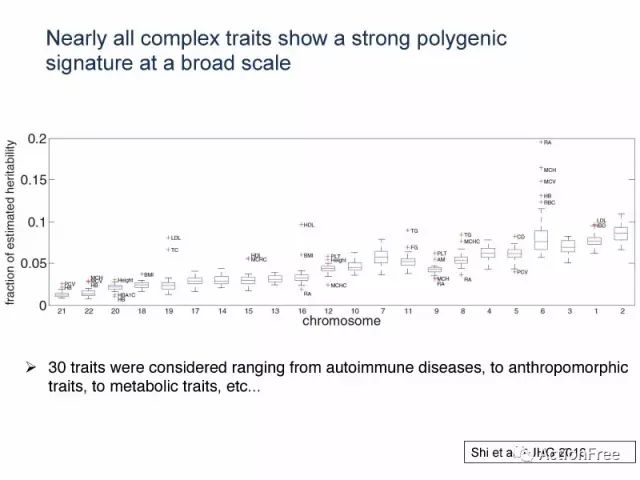
此外,研究者还观察了30种复杂性状,从自身免疫疾病,到代谢疾病等,发现几乎所有的复杂性状都在基因组上的很大的范围内呈现出非常强的多基因效应。
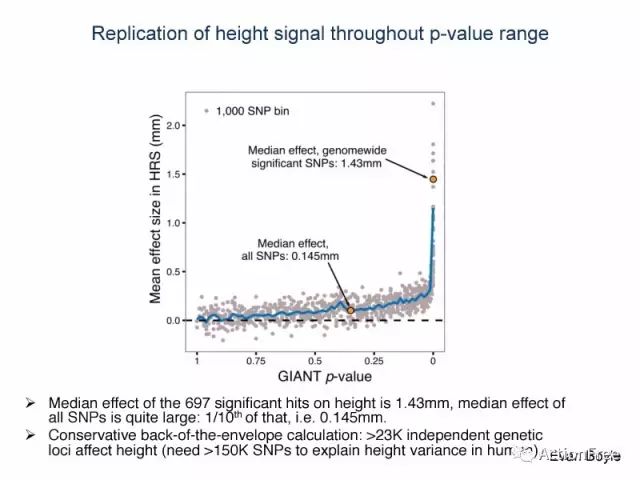
深入分析利用身高 ( height)的结果,此图首先利用GIANT项目25万人的基因组数据得到的与身高相关的位点的显著性水平进行排序(x轴,GIANT p value),评估他们在HRS(Health and Retirement Study)中对身高的效应的中位数(y轴)。
结论是GIANT显著的SNP在HRS项目的群体中所产生的效应的中位数是1.43mm,而基因组上的所有SNP所产生效应的中位数是显著位点的十分之一约0.143mm.
保守估计,大约需要大于150K的SNPs来解释人群中身高的差异,也就是说基因组上非常广泛的SNP都贡献了身高在人群中的差异。
观察1的结论是:对于很多性状来说,致病SNP几乎遍布整个基因组,而“疾病基因”却不是,说明致病SNP可能指向其他基因?
这是一场causal SNP 与 disease gene 的对决
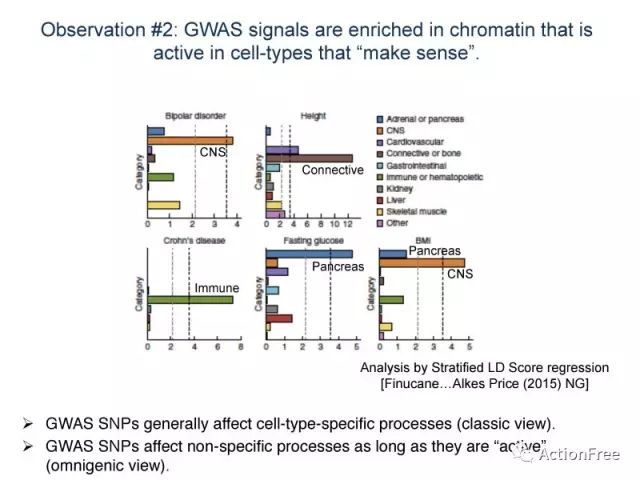
观察2: “不同疾病的 GWAS信号的富集具有细胞类型特异的活跃染色质区域 “是讲得通的。
图片解读: 该图来自Finucane等2015年发表在NG的文章Partitioning heritability by functional annotation using genome-wide association summary statistics. 这里只展示了6种性状的细胞类型特异性的富集,X轴表示的是富集分析的p值(−log10(P)),超过黑线说明显著富集,可以看到双向情感障碍信号富集与中枢神经系统,克罗恩病的信号富集于免疫系统,空腹血糖的信号富集于胰岛。此图支持 “不同的GWAS信号的富集,具有组织/细胞类型特异性”的观点。
与传统的观念相比:激烈的对比 ↓
传统的观念认为:不同疾病的GWAS信号的富集,具有细胞特异性;
泛基因组观念认为:只要这些区域是活跃的(有基因转录)那么GWAS信号就会对其产生影响。也就是不管这种染色质的状态是不是细胞类型特意的,只要active的就会受到影响。证据就是接下来的三张slides
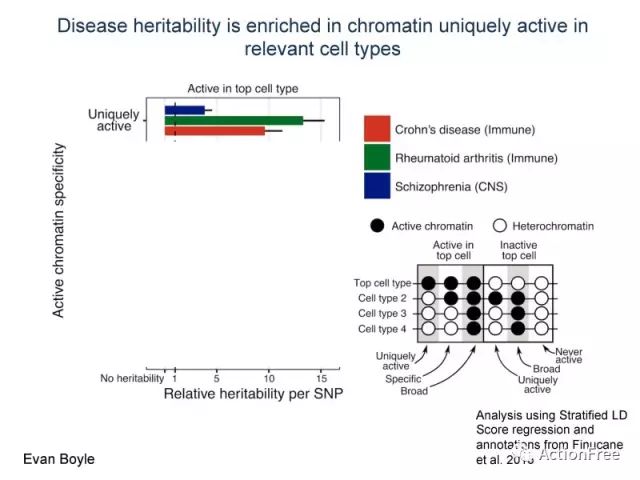
top cell type是GWAS信号富集的组织的细胞类型,在精神分裂症中指的是CNS,在克罗恩病中指的是免疫细胞,在类风湿性关节炎中指的是免疫细胞。根据右下角的图片,图中黑色的圆代表是 处于活跃状态的染色质(有基因转录的),白色的圆代表的是异染色质(高度浓缩的,不活跃的染色质),对于active 的部分,若只在top cell type 活跃,则为uniquely active chromatin, 若在不同的细胞类型均活跃,则为broad active chromatin, 若不同的细胞类型有差异,则为specific active chromatin.来区分uniquely active, specific active 和broad active. 对于uniquely active 的染色质,可以解释较多的遗传度,这支持了传统观念。然鹅~ 下图
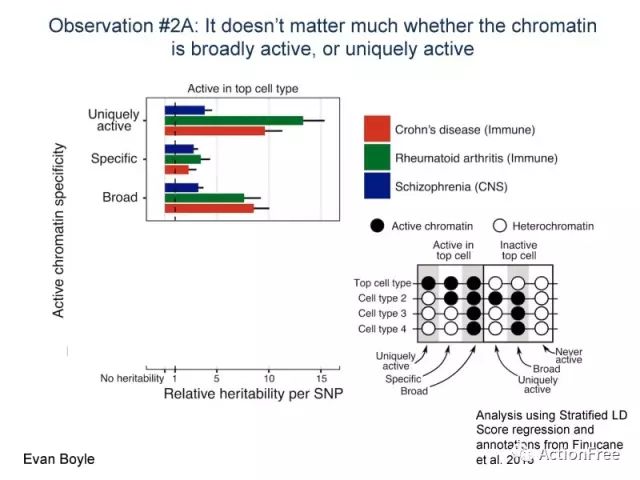
当我们观察broad active的染色质时,他们能解释的遗传度并没有比uniquely active 的染色质少,同时 specific active的染色质也参与解释遗传度。
因此观察2A的结论是 染色质到底是broad active还是sepcific active并不重要, 他们都会对遗传度有贡献,而且贡献也不必uniquely active的染色质少多少。而重要的是什么呢?是他们要active,接下来比较 intacive 的部分你就清楚了
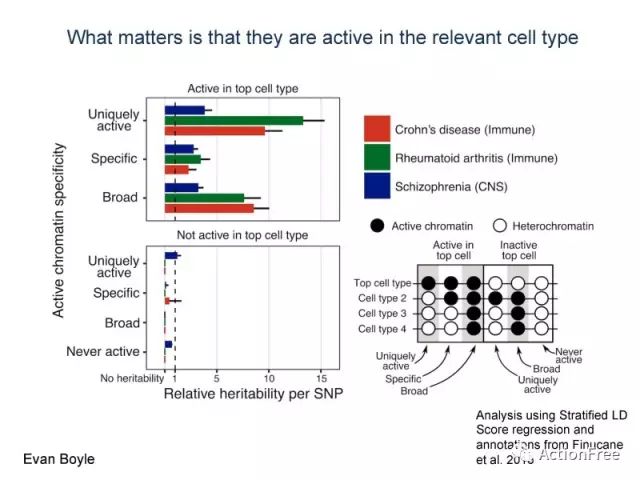
继续inactive的部分,这个部分的分类是按照右下角的图中,右边的部分来界定的,uniquely active是指在top cell type里面是异染色质在其他的一种细胞类型中活跃,broad active是指在除了在top cell type里面是异染色质外,在其他细胞类型都是活跃的,never active 是指在所有细胞类型都不活跃。这些在top cell type不活跃的染色质,贡献的遗传度,几乎没有(左下)。
这就说明,只要在top cell tpye里面活跃,不管你是怎么活跃,就会贡献遗传度;而若在top cell type里面不活跃,在其他的各种细胞都活跃也没有用。
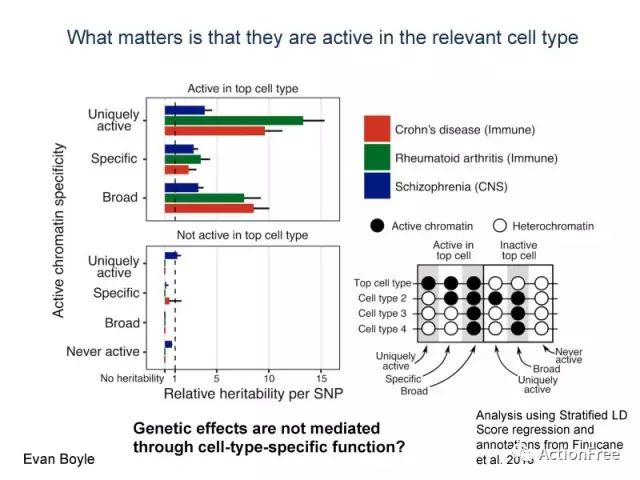
得出结论:遗传效应并不是通过细胞类型特异性的功能而介导的,而是只要在相关的细胞类型中活跃就会起到作用。
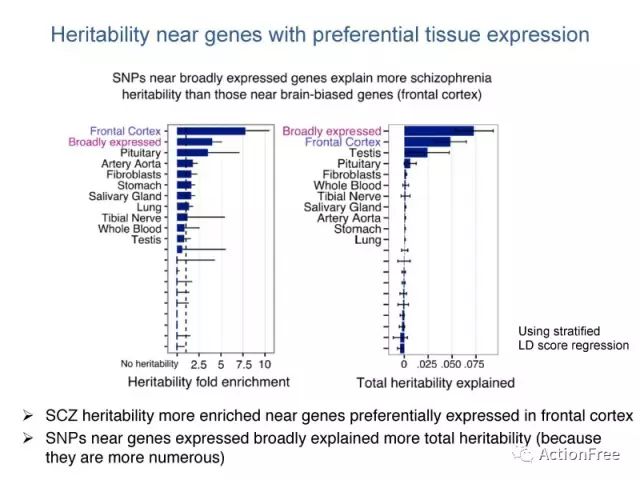
从基因表达的角度来看,以精神分裂症为例,左图显示,在前额叶皮质特异性表达的基因的遗传度的富集情况最高,而广泛表达的基因次之,而对于总体的遗传度的解释来看,广泛表达的基因能够解释的百分比更高,当然广泛表达的基因的数目更多也是一个重要原因。也就是说,一个基因不需要在这个组织中有特异的功能,而只需要他有功能,就会对疾病有影响。
所以研究者提出了如下假设↓

假设:那些在疾病相关的通路中没有直接的作用的基因,也可能对疾病有贡献,只要他们在疾病相关的组织中具有功能,这种功能不限于这个组织特异性的功能。
这个假设从何而来呢?有啥证据呢?请看观察3 ↓
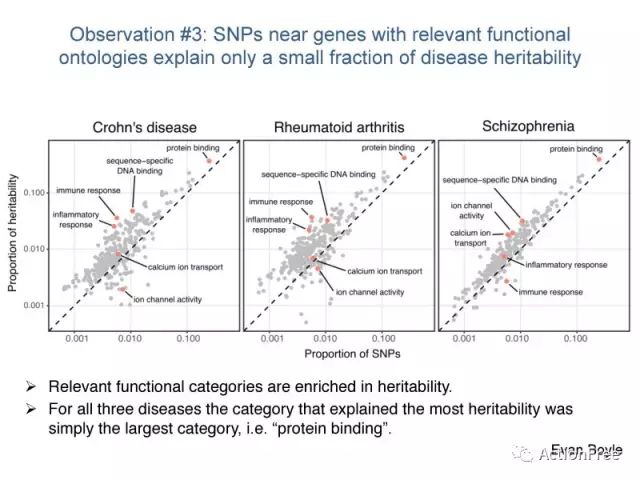
观察3:具有相关功能的SNPs附近的基因只解释了很小的遗传度。
图表解读:此图展示了三种疾病的基因功能的富集,x轴表示每一个功能条目中的SNP的比例,y轴表示对遗传度的解读。可以看到相关的功能(如克罗恩病相关的免疫应答,类风湿性关节炎的炎症反应,精神分裂症相关的钙离子运输等)所能解释的遗传度只有很小的部分,而解释最多的功能条目在三个疾病都是一样的——最大的条目蛋白质绑定。
这说明只看具有相关功能的SNP是远远不能掌握疾病的全貌的。

总结:基因组上大部分的变异都对遗传度有贡献。贡献遗传度的最主要的特点就是他们在相应的组织中活跃,而不需要作用于相应的通路。

根据如上的观察和假设,将基因分成三种类型:
核心基因:直接作用于疾病的基因,那些跟疾病相关的讲得通的基因,如精神分裂症中的突触相关基因;
外围基因:在相关的细胞类型中任何表达的或有功能的基因;
不活跃的基因:在相关的细胞类型不活跃的基因,处于异染色质状态的基因。
这些基因可以通过"小世界“网络(也叫无标度网络scale-free network)来发挥作用。

小世界网络的特点:大部分的点都能通过很少路径与其他任意的点连接。小世界网络被认为是最高效的网络。
熟悉WGCNA的小伙伴们,应该不会对小世界网络陌生。
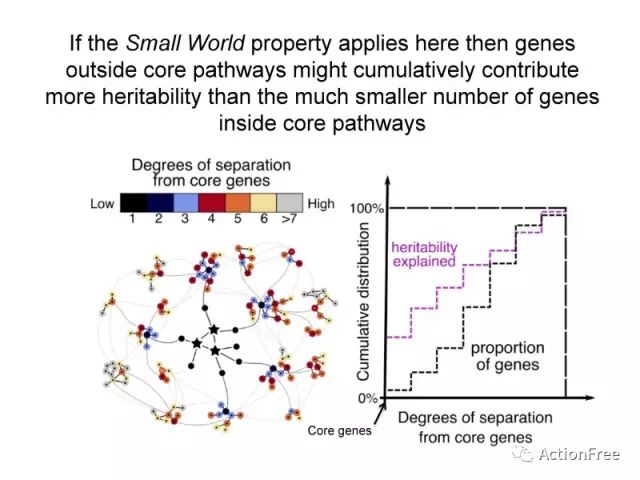
将小世界网络的特点应用于基因网络中,就会发现纳入那些核心通路外的基因(外围基因)将会增加对遗传度的解读。
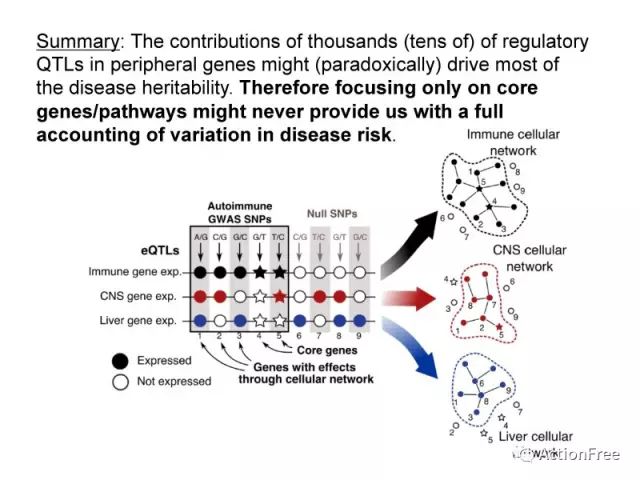
总结:数百上千的受QTL调控的外围基因可能贡献了主要的遗传度。因此只关注核心基因/通路将不可能完全解读变异对疾病发生风险的贡献。所以,有必要在泛基因模型的基础上来考虑复杂性状的遗传度问题。

对于复杂性状来说,小鼠模型的应用将非常受限,尤其是在失去功能的通路只贡献了疾病的很小部分的情况下;
药物的研发应该关注改变系统的状态,而不是只改变单个的基因或者通路;
加深对细胞类型特异性的基因/蛋白相互作用的理解。
 长按关注生信草堂
长按关注生信草堂












