参考湿疹血小板减少伴免疫缺陷综合征(WAS)的基因治疗临床试验,制定了符合药品生产质量管理规范(GMP)的慢病毒生产工艺。该工艺是基于瞬转细胞工厂中培养的HEK293F细胞。收集病毒上清可以达到50L,然后经过一系列膜及色谱纯化,收集水泡性口炎病毒糖蛋白型假病毒颗粒。该工艺可以将病毒浓缩200倍,并且杂质蛋白及DNA可以降低三个数量级。在六次大规模生产中,收集的病毒颗粒转染效率平均为13%。终产品中含有少量污染物,如来源于生产细胞的SV40大T抗原或宿主EIA序列。感染性病毒颗粒滴度高达2*109/ml。每批次可以生产6*1011个感染性病毒颗粒。纯化的WAS载体是具有生物学活性的,可以在WAS缺陷的B细胞系中插入的目的基因及高效表达并且可效转导CD34+细胞。以滴度为108个感染性病毒颗粒/ml感染CD34+细胞时,平均每个细胞含有0.3-1个载体考贝数,可以用于临床前应用。没有证据显示细胞学毒性。综上,来源于晚期HPV-1型病毒的慢病毒,其大量GMP制备、纯化及控制,是可行的。WAS载体的结果在初始小批量生产和质量控制有指导意义,有助于进一步研发及临床应用。
用于基因治疗的临床级逆转录病毒载体生产已经应用于一些病症,这种病毒载体修饰体细胞需要具有基因稳定性。γ逆转录病毒载体是首次应用于治疗严重联合免疫缺陷患者。如今,基于更加复杂的慢病毒设计的其他逆转录病毒基因转导载体,如人免疫缺陷病毒(HIV)-1,已经在临床上用于治疗遗传性疾病或HIV感染。作为整合型基因转导的工具,基于HIV设计的载体具有RNA基因组稳定性及转导非分裂细胞,优于γ逆转录病毒载体。多年以来,重组HIV(rHIV)病毒载体技术是安全的并且是非复制型基因转导系统,通过删除长未端重复(LTRs)的增强启动子序列,使病毒基因最小化,这样的设计可以防止插入基因组位置重组,防止失活病毒(SIN)再复制。在SIN载体中,可选择内源启动子调控目的基因,即通过内源启动子调控内源基因,同时启动目的基因表达,最小化插入突变的风险。
湿疹血小板减少伴免疫缺陷综合征(WAS)是采用源于HIV的慢病毒载体基因治疗的候选疾病。WAS是罕见的原发性免疫缺陷,其特点是血小板减少,复发性感染,湿疹。并且WAS具有较高的概率伴发自身免疫性疾病及淋巴系统恶性肿瘤。临床前试验表明体外基因转导自身造血干细胞是安全有效的。其采用的是第三代SIN rHIV-1来源的慢病毒载体转导,假病毒颗粒含水泡性口炎病毒糖蛋白(VSVg),内源性WAS启动子调控人WAS cDNA表达。对于治疗缺乏同种异体骨髓移植供体的严重WAS患者,这些结果为临床基因治疗研究奠定了理论基础。参考临床试验,本研究开发了符合GMP规范的大规模制备rHIV载体工艺。
据了解,几乎没有关于在GMP条件下生产rHIV的操作规范。在实践中,只批准一个批次的慢病毒载体应用于临床。rHIV临床载体生产方法目前主要是共转染2-5个质粒,这些质粒编码辅助性病毒蛋白或者目的基因。为了规避这种复杂工艺,已经报导一些稳定生产rHIV病毒的细胞系,但是都不符合GMP。因为293T细胞在生产rHIV方面的优势,所以仍然主要采用瞬转293T细胞的方法。所悉,生产rHIV病毒细胞系,没有与其他细胞进行比较的报导。一些衣壳糖蛋白可以用于包装rHIV假病毒颗粒,但是广泛应用的是水泡性口炎病毒G糖蛋白(VSVs),因为其宿主比较广泛。另外,从生产的角度而言,这种rHIV假病毒耐反复冻融,并且在浓缩和纯化时,具有良好的稳定性。因此适合在GMP条件下生产稳定、特征明确的产品。为了提高原始细胞转导效率,通常需要高浓度的rHIV颗粒,需要下游工艺浓缩病毒上清液。离心相对而言是粗鲁的浓缩方法,如通常采用的超速离心法,也用于离心细胞碎片,细胞膜组分,胞内及胞外(分泌至培养基)的蛋白。这些成分对靶细胞是有毒性的,尤其对原始细胞。如果应用于体内,可能引起炎症或免疫反应。因此除了浓缩,制备无毒性,成分更明确的rHIV颗粒纯化样品也是必要的。
本论文报导50L规模 rHIV生产,纯化采用常规的膜分离技术,离子交换色谱及凝胶过滤层析。该工艺生产的载体已经应用于WAS患者治疗。与科研级慢病毒载体制备相比,本文详述了在GMP条件下生产的批次特点,体外功能评价及临床应用中必不可少的安全参数。
HEK293细胞系来源于人胚胎肾细胞,是转染了截短的人腺病毒5型DNA片段,即转染了腺病毒E1A和E1B基因。Cell Genesys (South San Francisco, CA)提供一个HEK293细胞克隆(即293细胞)资助该研究。293T细胞系来源于HEK293细胞,是转染了一个质粒,编码SV40大T抗原(大T抗原是热不稳定的,并且转录由劳氏肉瘤病毒长未端重复调控),选择其中一个亚克隆,因为该亚克隆瞬转后具有高表达慢病毒载体的性能。该研究采用的293T细胞主细胞库及工作细胞库是GMP级别的。有些实验中采用293FT细胞(R700-07; Invitrogen,Cergy-Pontoise, France)。这些细胞都是来源于HEK293细胞(亚克隆为293F),是HEK293细胞(亚克隆为293F)经过编码SV40 LTA质粒转染的,SV40 LTA由CMV启动子调控。HCT116细胞用于病毒滴度测定,来源于人大肠癌,由American Type Culture Collection (CCL-247; ATCC, Manassas,VA)提供。293,293T,HCT116细胞系培养条件是37℃,5% CO2, DMEM( Invitrogen/GIBCO),10% FBS( HyClone, ogan, UT)。B淋巴细胞(B-LCL) 04W来源于WAS患者血液,该患者感染EB病毒(EBV)并已有报导(Charrier et al., 2007)。T淋巴细胞系8166-45来源于NIH AIDS Research & Reference Reagent Program (Germantown,MD),该细胞携带人类T淋巴细胞白血病病毒[HTLV]-1,但不表达。T淋巴细胞系C8166-45细胞和 B-LCL 04W细胞培养基为RPMI-1640,含10%FBS,谷氨酰胺及抗生素。
Zanta-Boussif及其同事报导的W1.6-huWASP-WPRE载体,其制备是通过瞬转293T细胞,由四个质粒组成:pRRLW1.6-huWASP-WPREmut6-K (或 pCCLW1.6-huWASP-WPREmut6-K),pKLgagpol(编码HIV-1 gag-pol基因), pKRev(编码HIV-1 rev基因),和 pKG(编码水泡性口炎病毒G糖蛋白)。所有载体都携带卡那抗性基因。在研发阶段,按照厂家(Macherey-Nagel, Hoerdt, France)建议,采用无内毒素的NucleoBond
PC 10000 EF千兆重力流柱。根据GMP要求,本次研究采用COBRA (Keele, UK).赞助的GMP级别质粒。
正前所述(Charrier et al.,2005).,一些研究中,对照质粒(编码eGFP的 质粒)及转移质粒apRRLPGK-eGFP-WPRE都是通过超速离心的方法进行纯化。
Nunc细胞工厂(十层细胞培养)进行大规模载体制备
在第0天,接种293T细胞于24个十层细胞培养的Nunc细胞工厂(CF10;Thermo Fisher Scientific, Waltham, MA)。细胞培养及生产用的培养基是含10% FBS(HyClone)的DMEM(GIBCO)。第三天进行细胞转染,转染前2小时进行细胞换液。根据文献(Grahamet al.,1977)报导,采用磷酸钙法进行转染,每个细胞工厂所用质粒量如下:pPGK-GFP or pW1.6-huWASP 760 μg , pKLgagpol 500 μg, pKG 270 μg, 及 pKRev 191 μg 。第四天(译者备注:转染后第一天)进行一次换液,收集转染后第24,48h培养上清。上清过滤除杂,然后核酸酶4℃过夜处理。核酸酶终浓度为5U/ml,商品名Benzonase,I 级,至少1.1*106U/mg,厂家Merck Chemicals, Darmstadt, Germany。DEAE离子交换色谱层析纯化病毒颗粒。750mM NaCl洗脱,收集洗脱液,含载体,通过中空纤维膜管束(nominal cutoff, 750 kDa; GE Healthcare Life Sciences,Orsay, France)渗滤至PBS中,再通过超滤法浓缩20-30倍,终体积约75ml。载体进一步经分子排阻色谱纯化,溶于X-VIVO 20培养基(Lonza, Basel, Switzerland)。将载体进行分装并储存于-80℃。
按照Charrier及其同事 (2007)所述,将一系列不同浓度的载体感染HCT116细胞后,通过qPCR法测定滴度。检测每毫升病毒上清中具有感染性基因组在HCT116细胞中的表达情况。按照Follenzi 和 Naldini (2002)所述方法测定慢病毒(LV)–eGFP滴度,采用HCT116细胞而不是Hela细胞,将滴度定义为转导单位/毫升(TU/ml)。物理颗粒滴度通过ELISA法(HIV-1 p24 core kit, NEK050B; PerkinElmer Life Sciences, Boston, MA).测定衣壳蛋白(p24)。
光谱法测定总蛋白浓度,即采用Bradford法,灵敏度达10 μg/ml。荧光法测定总DNA浓度,即采用PicoGreen法 (Invitrogen)。灵敏度达1 ng/ml。ELISA (Merck)法测定核酸酶含量,灵敏度达200pg/ml。ELISA法测定宿主蛋白含量,采用HEK-293 HCP 试剂盒(Cygnus,Technologies, Southport, NC),灵敏度达300 pg/ml。ELISA法测定BSA含量,灵敏度达250 pg/ml (Cygnus Technologies)。
复制型慢病毒(RCL)分析方法见Escarpe 及同事所述。阳性对照是共转染野生型HIV-1型前病毒质粒和编码衣壳糖蛋白的质粒。其中,前病毒质粒缺少辅助性病毒基因(DvifDvprDvpuDnef),编码衣壳糖蛋白的质粒必须与生产假病毒颗粒的载体一致。根据报导(Escarpe et al., 2003),结合VSVg假病毒载体生产实际情况,推测该分析方法的灵敏度约是,每T-175培养瓶含有的半数组织培养感染剂量(TCID50)是2*108个转导单位。
载体中E1A和SV40大T抗原序列是采用qPCR法,分别特异性扩增64bp E1A序列和112 bp SV40大T抗原序列。E1A和SV40大T抗原序列测定实验中,在提取之前,均加入内参。量化内参可以提高DNA提取效率并使PCR扩增标准化。E1A和SV40大T抗原序列测定定量的限度是5000 考贝/ml。
质粒DNA(VSVg)、腺病毒(E1A and E1B)、SV40(大T抗原)基因组序列转染靶细胞
将载体转染C8166-45细胞后,细胞扩增共计29天,六个重复。DNA污染物的的测定是采用qPCR法。检测VSVg时,采用pMD.G质粒制作标准曲线。检测E1A和SV40大 T抗原时,采用pTOPO_ALB_E1A 和 pTOPO_ALB_AgTSV40质粒制作标准曲线。pTOPO_ALB_E1A 和 pTOPO_ALB_AgTSV40质粒包含一个细胞蛋白基因(白蛋白)及一个考贝E1A或一个考贝SV40大T抗原。100ng基因组DNA中特异性序列,根据实验的不同,检测限度从20-1000考贝不等。
脐带血(UCB)袓细胞CD34+细胞通过免疫磁珠法筛选(Miltenyi Biotec,Paris, France)得到。在0.2ml X-VIVO 20培养基中过夜孵育5*104个CD34+细胞(Lonza),还添加有青霉素(50 U/ml),链霉素(50 mg/ml),2mM L-谷氨酰铵(GIBCO-BRL/Invitrogen),干细胞因子(50 ng/ml),Flt-3配体(25 ng/ml),血小板生成素(25 ng/ml), 白介素-3(10 ng/ml) (均来源于 Preprotech, Neuilly-sur-Seine, France)。慢病毒载体转导已活化细胞6小时,浓度从107至108 IG/ml,同时需要加入海美溴铵(Polybrene, 6 mg/ml;Sigma-Aldrich, St. Quentin Fallavier, France)。第二天,洗涤细胞,并通过两个不同的方法检测细胞是否发生分化:克隆形成单位比例(CFU-C)或通过qPCR法测定细胞数量、活率测定细胞扩增和转导。CFU-C是采用在MethoCult 培养基 (H4434;StemCell Technologies, Vancouver, BC, CA)中铺板1000个/毫升转导或未转导细胞。两周后,在倒置显微镜下检测红细胞克隆形成单位比例(CFU-E)、粒细胞-单核细胞克隆形成单位比例(CFU-GM)及混合细胞克隆形成单位比例(CFU-mix)。在扩增培养基中,转导用CD34+细胞接种于24孔平板中,用X-VIVO 20培养基将细胞密度调整为5*104个/ml,培养基中还需要添加血清、抗生素、谷氨酰铵及细胞因子。在37℃ 5% CO2中培养8天,每三天需要补加新鲜培养基。培养结束,台盼蓝染色法测定细胞活力及Wizard genomic DNA purification kit (Promega, Madison, WI).提取基因组DNA。参考文献报导(Charrier et al., 2007),采用qPCR法测定每个细胞中载体整合考贝数,通过测定土拨鼠肝炎病毒转录后调控元件(WPRE)载体序列校正人白蛋白基因。
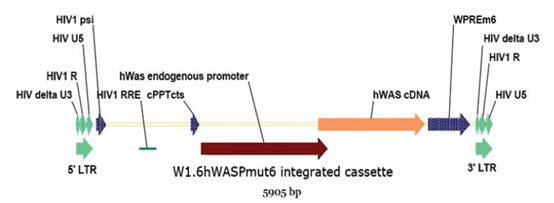
FIG.1 WAS载体整合进靶细胞后的前病毒序列示意图。HIV,人类免疫缺陷病毒。Psi,衣壳包装序列。PBS,引物结合位点序列。LTR,长末端重复序列,5’或3’端。HIV R,HIV R区。HIV delta U3,经过删除的U3区带。RRE,Rev反应元件。cPPT cts,中央聚嘌呤—中央末端序列。hWAS,人WAS基因。WPRE m6,突变的土拨鼠肝炎病毒转录后调控元件(Zanta-Boussif et al., 2009)。彩图来源网址
www.liebertonline.com/hum
.。
载体假病毒颗粒感染WAS蛋白阴性细胞B-LCL条件是:0.2ml/孔,2.0*105个细胞,加入5-二氮十一亚甲基聚甲溴化物(6 mg/ml)处理6小时。载体浓度变化从0.05 至10_107 IG/ml,感染复数(MOI)变化从0.5至450。转导后,洗涤细胞并培养8-10天,每三天添加新鲜培养基一次。WASP的表达检测按照文献(Charrier et al., 2007)采用免疫印迹法。
VSVg假病毒WAS慢病毒载体,已经有报导( Charrier et al., 2007; Galy et al., 2008),开发用于体外转导患者自身CD34+细胞,引导基因组整合5.9kb表达表达元件,表达WAS蛋白(Fig. 1)。从生产用于临床研究的角度,这种载体生产规模按照如下参数推断。考虑到20Kg年轻患者接受5*106 CD34+细胞/Kg,MOI固定为50。那么每名患者约需要感染载体5*109假病毒颗粒。为了制备一批载体用于治疗数名患者(至少5名),考虑到质量保证及法规,GMP 质量控制将消耗一批中的一半,因此每个批次至少生产5*1010个rHIV病毒颗粒。按照经验(Kutner et al., 2009),转染293T细胞后,每毫升可以产生约107个具有感染力的VSVg假病毒颗粒包装的rHIV 载体。考虑到纯化及浓缩步骤产率约10%,估计每个批次需要50L病毒上清液。这种规模的生产可以采用10层细胞工厂(CF10)培养生产细胞,病毒上清生产流程是在闭路完成的,可以保证符合GMP(Przybylowskiet al., 2006)。此外,从组织培养过程而言,这些容器直接放大转染环境,与表面积成正比。
WAS–LV载体生产是通过共转染四个质粒,分别单独编码HIV-1 Gag-Pol病毒蛋白及酶,HIV-1 Rev蛋白,VSVg假病毒,WAS基因。因为WAS载体是新一代载体,所以其基因转导元件删除了所有HIV辅助蛋白序列及tat。混合的5’LTR使Tat不依赖病毒RNA的产生。混合的5’LTR制备,可以通过劳氏肉瘤病毒(RSV)或CMV启动子与HIV R-U5元件融合,分别构建成pRRL-或 pCCL型。为了评估构建的5’LTR对WAS载体的影响,比较了小量四质粒转染后的滴度。四质粒系统是pRRL-W-WAS 或 pCCL-W-WAS,只有5’LTR是不同的。超速离心后测定效价。小量生产是在T-225培养瓶中完成的,pCCL比pRRL滴度高两倍[1.47 (±0.57)*1010 vs.0.84 (±2.2)*1010 IG/ml],并且病毒颗粒具有较高的感染力[1.8 (±0.6)*105 vs. 1.1 (±0.1)*105 IG/ng p24]。在CF10培养瓶中也得到相似的结果(5.5*1010vs. 1.6*1010 IG/ml and 1.4*105 vs. 4.4*104 IG/ng p24)。这些结论与先前报导(Dull et al., 1998)一致,所以本研究选择pCCL转染载体。
HEK293细胞及其衍生细胞293T均瞬转用于生产慢病毒。虽然293T细胞相比其HEK293细胞应用更广泛,但作为母本的HEK293细胞在安全方面更具优势,因为HEK293细胞缺少SV40大T抗原(SV40 LTA)编码基因,该基因属于癌基因。因此构建eGFP,在T-75瓶或CF10瓶中小量生产,比较HEK293细胞和293T细胞生产慢病毒的能力,进行快速评估。正如预期,293T细胞比HEK293细胞明显复制周期短(19 vs. 25 hr)。转染24小时后,293T细胞中eGFP+细胞明显多于HEK293细胞,表明293T细胞具有较高的转染效率(结果未显示)。在未经优化的条件下,293T细胞比HEK293细胞病毒上清效价更高(293T细胞,在T-75系统1.2*107 ,在CF10系统 3.7*106 TU/ml 。HEK293细胞,在T-75系统2*106,在CF10系统9.9*105TU/ml)。与受青睐的293T细胞相比,HEK293细胞在CF10系统中未能充分提高病毒生产或滴度。在最优条件下采用eGFP,293T细胞所需载体总量比HEK293细胞高四倍(1.74*1010 vs. 4.59*109 TU)。此外,就载体转染效率而言,293T细胞比HEK293细胞高两倍。这些结果,使得慢病毒生产工艺研发采用293T细胞。细胞中出现SV40大T抗原这种特殊污染物,需要在载体生产中监控其含量。为了生产临床级载体,293T细胞工作细胞库是在GMP环境中制备的并且得到认证。
rHIV慢病毒颗粒生产工艺流水线操作是为了减少操作步骤及促进工艺符合GMP要求。公开的操作规范基于磷酸钙法转染293T细胞并且只收集两次(每天一次)就可以扩大到24个CF10培养瓶,可以生产48-50升病毒上清。生产WAS载体的质粒浓度与数量是在一个瓶中进行优化的,放大是在相应的24个CF10培养瓶进行的。根据其他公开操作规程,WAS转移质粒浓度是超过其他质粒浓度的。矩阵实验中,四质粒系统中每一个质粒浓度都是变化的。变动设置值的0.5至3倍,对WAS LV滴度没有影响,例外是VSVg质粒量大时滴度有下降,这与其他人的实验结果一致。采用相同的工艺流程大规模制备六批WAS载体,得到的原始病毒上清液效价范围是1-5*107 IG/ml,优质病毒颗粒感染力范围从0.4*105至1.4*105IG每钠克P24(Table 1)。但这些病毒上清液也含有污染物。在研发阶段四个实验当中,初始总蛋白浓度是6.14±1.47 mg/ml,最初总DNA浓度是1.97±0.0 mg/ml。因此在启始物料中,每108IG,蛋白浓度平均值是21.4mg,DNA浓度平均值是6.9 mg。
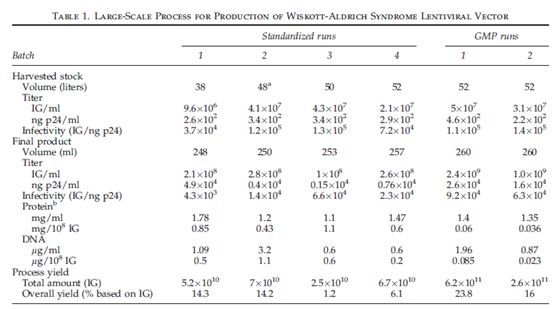
IG,感染性基因组。GMP,良好操作规范。
a 处理12升。
b 每毫升X-VIVO 20培养基中含1.12mg蛋白质。
下游工艺操作规程正如其目的:病毒载体的纯化和浓缩,去除工艺及细胞来源的杂质,最终制成临床级载体制剂。在临床前研究中,当使用WAS慢病毒载体,浓度为0.5-1*108IG/ml时,WAS表型具有较高正确率。因此本研究计划生产载体终浓度至少为2*108IG/ml。从GMP应用的角度,结合自己经验及先前发表的逆转录病毒或慢病毒纯化研究,采用多步纯化及浓缩。我们自身经验是一些色谱法及基于膜的工艺步骤具有优势。该工艺成功应用于六批大规模质粒生产(Table 1)。其中四批是在研发阶段的标准条件下制备的。最后两批因为计划用于临床,所以按照GMP生产,与另外四批使用的细胞与质粒是完全一样的,不同的只是最后两批的生产使用的材料是GMP级别的。
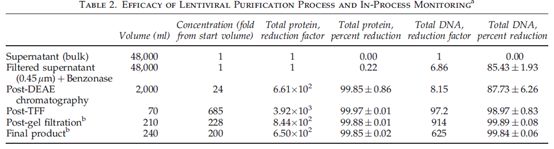
第一步,病毒上清液通过降低孔径的低截留膜过滤器(0.8um降至0.45um)预处理。然后,滤过液经核酸内切酶(Benzonase) 4℃过夜,以去除质粒及其他DNA污染物。具有感染力的病毒颗粒,收集到约三分之二,经过膜处理和DNA酶处理,感染力下降不足2倍(Fig. 2B),降低了产品85%的DNA含量(Table 2)。阴离子交换色谱法是纯化逆转录病毒非常有效的方法,也采用该方法捕获病毒颗粒。病毒上清液通过DEAE离子交换色谱柱纯化,PBS洗涤、2L氯化钠洗脱。该工艺步骤去除99%以上的总蛋白质(Table 2)。洗脱液通过切向流过滤去盐并浓缩约20至30倍,VSVG-假型慢病毒颗粒具有较高的产量。在这一步,通过中空纤维管束产生约70ml产品。进一步减少蛋白质和DNA污染物(Table 2)。最后,浓缩病毒悬液通过分子排阻色谱法(载样体积:柱体积=0.15)置换至制剂培养基(X-VIVO 20)。约200ml收集液通过0.2um胶囊式过滤器除菌,过滤器再经约40-50ml 载体制剂培养基清洗,终产品体积约250ml。这步工艺,体积浓缩了200倍,总蛋白和DNA污染物减少了三个数量级,每毫升残留蛋白量为1.38±0.3mg,每毫升残留DNA量为1.38_1±24 ug(Table 2)。下游工艺将总DNA量降至0.6–3.2 ug/ml,最后108 IG含DNA 0.023–1.1 ug (Table 1)。去除DNA最有效的步骤是核酸酶处理(去除85.4±1.93%)和切向流过滤(去除14.5%),去除总DNA的98.97±0.83%。去除蛋白最有效的步骤是DEAE色谱法,去除了99.85±0.09%的总蛋白。总蛋白约去除了700倍(Table 2)。下游步骤没有明显的去除效果,因为因为制剂培养基中含有蛋白质。在分子排阻步骤中,载体制剂使用的X-VIVO 20培养基所含蛋白量大于1.12mg/ml,因此残留蛋白水平在最终产品中有所提高。在研发阶段,每毫升终产品中含有蛋白1.38±0.30mg(每108 IG含蛋白0.74±0.29mg)(Table 2),在GMP条件下,每毫升终产品中含有蛋白1.35和1.4 mg(每108 IG含蛋白不足0.1mg)(Table 1)。当考虑添加X-VIVO 20培养基,并扣除培养基中蛋白,可以估算出,每108 IG含蛋白0.12±0.14mg(Table 1)。考虑到最初病毒上清液中,每108 IG中有21mg 蛋白质,这些数据表明该工艺可以有效去除蛋白质,使病毒颗粒得以纯化。无论是否统计最后制剂培养基中的蛋白质,两批次终产品中,每108 IG,分别含蛋白0.74±0.29 或 0.12±0.14mg。远小于Schonely及其同事建议的蛋白质污染含量(每108 IG中蛋白质含量小于7mg )。
综上,六批大规模生产,该工艺达到最初规定的2*108 IG/ml,病毒颗粒的感染力大于1*104 IG/ng p24。但总产量不高,只有10%。每批次可以生产高达5*1010 IG,因此可以达到最初规定。然而,GMP条件下生产的两批产量超过最初规定,较高载体效价(>109 IG/ml),高的感染力(>4*104 IG/ng p24),较高产率(16–23%),每批可以产生2–6*1011 IG(Table 1)。
在工艺当中,最重要的浓缩是离子交换色谱及超速离心,分别提高感染力67±23倍和163±113倍,这两种方法在研发及GMP生产都会涉及到(Fig. 2A)。在相应的步骤中,物理颗粒滴度也在增加。在最后步骤中,发现效价降低,可能是载体稀释及载体轻微吸付到除菌膜。阴离子交换色谱纯化后感染力有所降低(IG/ng P24 比值),在超速离心步骤时感染力有所回升。这种原因可能是柱洗脱时需要增加盐浓度,盐可能影响qPCR法检测感染滴度,但不影响ELISA法检测物理滴度。确实,下游的DEAE色谱法中,感染性基因组与纳克P24蛋白比值有所提高(Fig. 2B)。研发与GMP生产有两外不同:第一,工艺开始时,GMP生产具有较高的病毒上清滴度。第二,GMP生产比研发而言,切向流过滤收率稍高(Fig. 2A)。综上,初始体积与终产品比值,研发阶段的载体(感染力)质量下降40%,GMP生产当中下游工艺保持病毒颗粒感染力相当稳定 (Fig. 2B)。
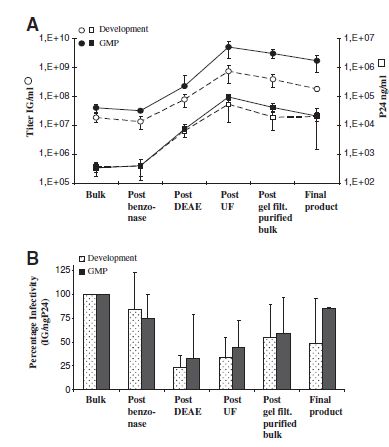
FIG.2 研发阶段大规模生产及GMP生产下游工艺步骤分析。(A)在研发(虚线)或GMP生产(实线)中WAS慢病毒载体生产工艺各环节,平均感染滴度(IG/ml; 圆圈)和P24滴度(ng p24/ml; 方形)。(B)颗粒感染力下游工艺各步骤中以IG/ng p24表示,计算与初始病毒上清百分比。
点柱图:研发。实柱图:GMP生产。IG,感染性基因组,UF,超滤。
TFF,切向流过滤。
a 第二、三列标出了每步工艺中理论体积与浓度。研发阶段四批大规模生产中蛋白质和DNA含量,是通过测量实际加工与收集产品的体积,计算去除效率。平均启始总蛋白浓度是6.14±1.47 mg/ml,平均启始DNA浓度是1.97±0.0 ug/ml,最终总蛋白浓度是1.38±0.3 mg/ml(包括制剂培养基中加入的蛋白量),最终总DNA浓度是1.38±1.24 ug/ml。
b X-VIVO 20制剂培养基蛋白质浓度是1.12mg/ml。
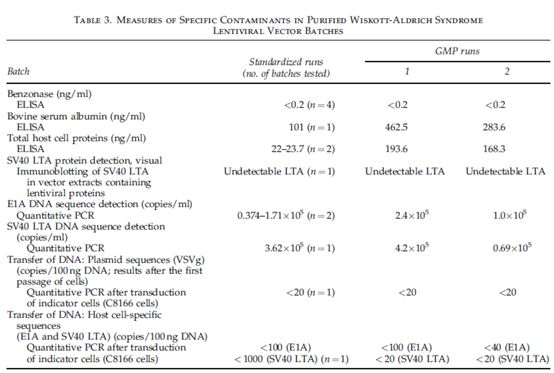
在工艺控制过程中测量,发现下游工艺分别减少总DNA 1000倍及蛋白量700倍(Table 2)。为了促进终产品转导造血干细胞,因此终产品溶解于 X-VIVO 20 培养基,每毫升培养基中约含1.1mg蛋白。生产培养基,生产细胞,或者工艺也可能出现其他蛋白污染物,可能对患者使用产品造成一定的危险。因此在研发阶段检测特定污染物,为GMP生产提供参考。工艺副产物之一是核酸酶Benzonase,在工艺启始以5ng/ml的浓度与病毒上清共孵育,因此相当于每个批次额外加入了2.5*105单位核酸酶。这种外源性酶,是载体生物活性不需要的,在工艺下游是要去除的。最终的纯化与制剂产品,不论研发还是GMP生产,都是检测不到的核酸酶的(Table 3)。牛血清白蛋白在病毒生产培养基中常用浓度是2.5mg/ml,终产品中其浓度不足500ng/ml,表明该蛋白质去除效率至少5000倍(Table 3)。源于生产细胞的总宿主蛋白检测是通过HEK293特异性ELISA实验,发现有少量残留,未超过200ng/ml。
生产细胞的特异产物SV40大T抗原(SV40 LTA),在许多细胞中都有致癌性。病毒上清超速离心后采用western blot,可以检测到浓缩慢病毒载体中SV40 LTA的残留(Fig. 3).。相反,同样采用western blot,载体纯化采用多步膜过滤和色谱法,未检测到SV40 LTA残留。所有纯化或超速离心样品含有等量的病毒颗粒,见HIV p24 和 p17免疫印迹图谱(Fig. 3),表明纯化工艺可以去除该特异污染物。为了进一步评估产品致癌性,检测了E1A 或SV40 LTA DNA序列含量,这些可能从生产细胞的培养过程中释放。通过定量PCR,检测到每毫升纯化载体中,检测到E1A含量是0.37-2.4*105考贝,SV40 LTA含量是 0.69-4.2*105考贝。平均每个293T细胞含E1A 6.6考贝,SV40 LTA 2.1考贝。考虑到293T细胞中DNA C值为9.8ng(认为是三倍体细胞),两批GMP生产的载体,包含0.05-2ug基因组DNA/ml。代表着高达70%总DNA(荧光法检测)(Table 1)。残留DNA可能来源于转染细胞用的质粒,为了评估E1A 和 SV40LTA 序列的意义,我们验证了具体多少量可以转染靶细胞并导致感染。本研究用培养敏感型指示细胞(C8166-45)时加入载体并扩增细胞。第一个检测点是转染后一周,通过qPCR未检测到E1A和SV40 LTA序列信号。因此,载体中少量残留E1A和SV40 LTA序列不会转染靶细胞。该实验中也检测了其他可能转染DNA序列(如来源于质粒的序列)。未检测到指示细胞中的VSVg序列(来源于质粒)(Table 3),然而,超速离心制备样品转染靶细胞一周后,可以将这些序列转染进靶细胞(数据未显示)。
GMP,药品生产质量管理规范。 LTA,大T抗原。 SV40,猿猴病毒40. VSVg 水泡性口炎病毒糖蛋白
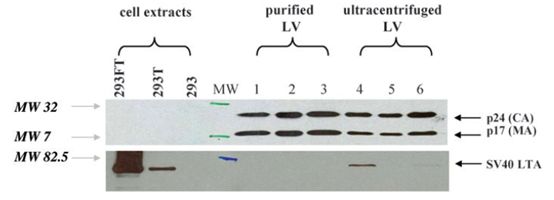
FIG. 3. 慢病毒载体中病毒性蛋白SV40大T抗原检测与鉴定。免疫印迹法中,抗体特异性检测HIV p24衣壳蛋白和p17基质蛋白(如图所示,上面条带标出蛋白24KDa与17KDa,marker标示出32KDa与7KDa)及SV40大T抗原(如图所示,下面条带,蛋白分子量80–95KDa,marker标示出82.5KDa)。1-3号泳道表示研发或GMP生产中rHIVWAS慢病毒载体纯化与生产的一些样品。4-6号泳道表示WAS 或GFP rHIV 载体小规模制备并经超速离心浓缩。293 和293T细胞提取物分别做为阴性和阳性对照。293FT细胞因为高表达SV40大T抗原,也做了检测。
CA,衣壳。 LTA,大T抗原。LV,慢病毒。MA,基质。MW,分子量。彩图网络链接
www.liebertonline.com/hum
。
尽管慢病毒载体系统有许多安全问题,但必须在许可条件下使用灵敏的方法证明无复制型重组HIV颗粒(复制型慢病毒,RCL)。六批WAS慢病毒载体生产中无RCL,六批生产包括三批小规模,研发阶段一批大规模,两批大规模GMP生产。每个批次中,总体积的5%使用p24衰减法检测,如上所述(Escarpe et al., 2003)。阳性对照是缺乏辅助基因的HIV-1弱毒株,但编码HIV和VSVg衣壳,TCID50 值相当于10-20 fg的p24蛋白。脉冲控制表明,采用该对照,任何批次WAS 没有抑制RCL检测。六批次检测WAS 慢病毒,C8166-45细胞扩增28天后,p24蛋白低于检测限。两批GMP生产批次,检测了感染性颗粒的数量,结果表明,1.25–3*1010 IG中,少于一个RCL,考虑用于临床试验的剂量,这符合监管要求的每名患者少于一个RCL。










