
读了《现代液相色谱技术导论》,我受益匪浅,分享下我的读书笔记和我读这个书的精彩内容。
一:
HPLC分离模式
|
色谱模式
|
色谱柱极性
|
流动相
|
样品
|
|
反相色谱法
RPC
|
非极性(
C18
)
|
极性:水和有机溶剂的混合液
|
水溶性样品
|
|
正相色谱法
RPC
|
极性(未键合硅胶颗粒)
|
极性较弱的有机溶剂混合相(如乙烷、二氯甲烷)
|
不溶于水的样品、制备性
HPLC
、异构体
|
|
非水反相色谱法(
NARP
)
|
非极性(
C18
)
|
有机溶剂的混合溶液(乙腈加二氯甲烷)
|
疏水性很强,不溶于水的样品
|
|
亲水作用色谱(
HILIC
)
|
极性(硅胶或酰胺
-
键合相)
|
水和有机溶剂(如乙腈)混合溶液
|
高度极性的样品,不容易在反相色谱保留
|
|
离子交换色谱(
IEC
))
|
色谱柱含有带电官能团,可以键合带相反电荷的样品离子
|
盐的水溶液和缓冲盐,
|
可电离的样品,分析生物大分子
|
|
离子对色谱(
IPC
)
|
非极性(
C18
)
|
流动性加入带相反电荷样品的离子作用的离子对试剂
|
反相色谱中保留较弱的酸或碱
|
|
尺寸排阻色谱法(
SEC
)
|
惰性的色谱柱
|
水或有机相
|
基于样品分子大小分离;分离分子大分子或者合成聚合物
|
二:色谱分离的过程
1.基本概念
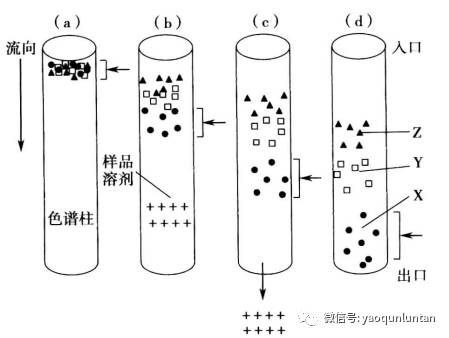
假定含有三种不同化合物分离,
●
代表化合物
X,
▲
代表化合物
Y,
□
代表化合物
Z。如图中没有显示流动相分析,溶解样品的溶剂分子用+代替。当样品进样到图a的色谱柱上,有流动相带动,在不同阶段内留个色谱柱,然后离开色谱柱进入检测器如图e
,并生成一个检测器响应对应时间作图的结果(色谱图)。当样品在色谱柱中(图
a~d),样品XYZ分子展现出两种行为:差异移动和分子扩散。
当一个给定的溶质分子通过色谱柱时,这些分子迅速扩散,占据色谱柱中大部分体积。
给定溶质分子在色谱柱里所占的体积定义为色谱带。
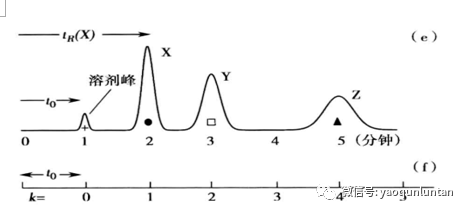
当色谱带离开色谱柱时,它就在色谱图中被记录下来,为色谱峰。
每个色谱峰的身份可以由他们通过色谱柱的时间来定义(保留时间)。保留时间是指样品进样后到出现色谱峰顶端时的时间段。
溶剂峰的保留时间是色谱柱的死时间。
保留因子
k:固定相中的溶质分子总量除以流动相中溶质分子总量。
或者
其中:
t
R:
保留时间
t
0:
死时间
其中:
t
0
=Vm/F,F为流速ml/min,Vm为色谱柱内流动相总和
。
Vm≈5*10
-4
Ld
2
L
:柱长
mm;d:内径mm
2.分离条件和样品组成的角色
反相色谱法中使用非极性的色谱柱和极性的含水的流动相。极性的溶质分子喜欢极性的流动相(物以类聚)而保留程度较低(较大的
R值,较小的K值),而非极性的溶质分子会倾向和非极性的固定相相互作用并能保持更久。反相色谱柱的流动相通常是由水或水性缓冲盐(A相)和有机溶剂(B相)组成。当有机溶剂增加时,所有样品化合物的保留都会减少。%B值改变+10%,K值一般会减少到原来的1/2或1/3.
3.分离度和方法建立
HPLC方法的建立:选择合适的分离条件,以便为一个给定的样品提供可靠的分离结果。
当需要分离多个色谱峰时,对于分离程度最差的一对色谱峰来说,分离度的最低要求是
Rs≥2,这样的色谱峰对称为决定性的色谱峰,分离度为决定性的分离度。
分离度
Rs=(1/4)[k/(1+k)](α-1)/N
0.5
(
a) (b) (c)
分离度:与保留因子
K;分离因子α和理论塔板数N有关。提高
分离因子
α是最有效的方法。
3.1优化保留因子k
在等度洗脱中,
k可以通过改变%B值来控制。
通常把所有峰的分离目标设置在
1≤K≤10(或者法规部门要求k≥2).以下列来说明。
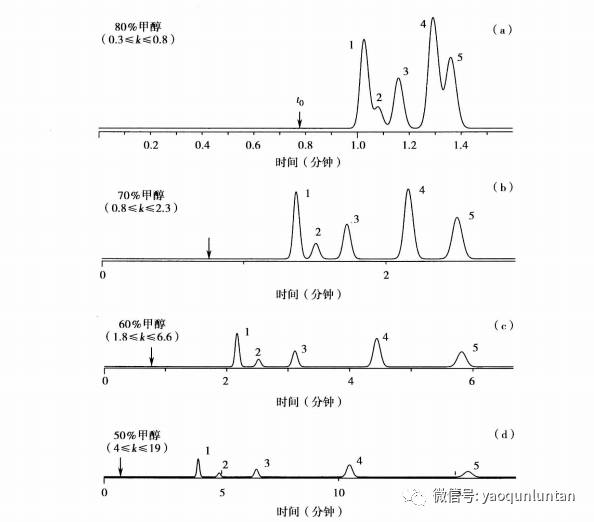
首先,
80%B或者90%B来展开最初的分离,保证样品全部洗脱。如a所示0.3≤k≤0.8,根据每减少10%B,k值增加2.5倍规格,当为70%B时,第一个风的k为2.5*0.3≈0.8.最后一个为2.5*0.8≈2.0,即0.8≤k≤2.0.当60%B时,为2.0≤k≤5.因此可使用60%B来实验。
当样品保留程度太低,
80%B时开始所有的峰k≈0(即一个单一峰包含所有峰),可以是降低30%B即用50%B开始实验,然后再进一步改变B%值得到合适的k值。由于2.5倍法则只给予的是近似值,因此最好不要在不同的实验间对流动相B%改变大于30,防止样品的丢失。
3.2 优化
选择性
α
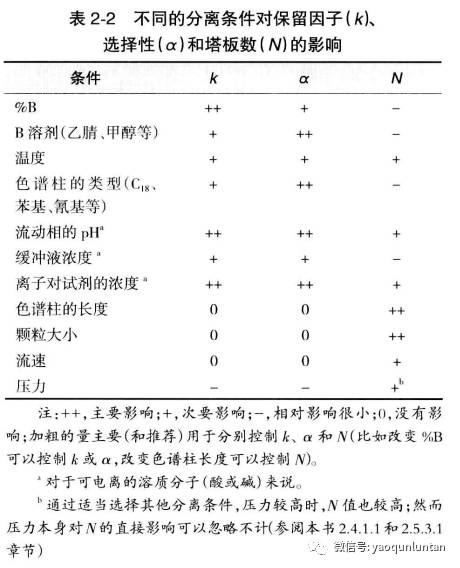
通过
2-2的表中可看出:%B;B溶剂的选择;温度;柱子类型;流动相PH;缓冲盐浓度;离子对试剂浓度都可以改变分离效果。
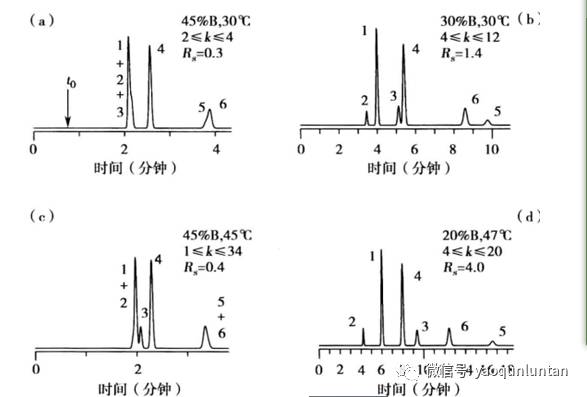
上图为例:
a图中,条件:45%B和30℃,1-3和5-6的峰分离度较差,
b图中,条件:30%B和30℃,1-3和5-6的峰分离度较好,3-4分离不够。5与6 的位置对调。通过观察可以看出适中的B能使3-4分开(在峰对1.2和峰对5.6接近之前)
c图中,条件:45%B和45℃,与a比。
d图中,条件:同时改变B和温度。
为了获得可接受的分离度效果,重心应该放在选择性的改变上(主要是
B%和温度)。
3.3 优化色谱柱的塔板数
通常增加色谱柱长度能增加塔板数
三、方法的建立
分析方法的建立包含步骤:
样品的组成和分离目标的评估
样品的预处理
色谱模式的选择
检测器的选择
分离条件的选择
潜在问题的评估、识别和解决之道
方法的确认和系统适应标准的确定
1.样品的组成和分离目标的评估
在建立方法前应该对样品有一定的了解,如酸碱性、
pka等,如果有酸性或者碱性化合物,应该控制ph值,以及考虑pka值等。样品的分子量影响分离条件的选择,等等。
2、样品的预处理
可能需要去除损害柱子或者干扰目标的成分。可以通过:减小样品的大小;干燥;过滤;液液萃取;固相萃取;衍生化等方式来进行。
3、色谱模式
RPC是HPLC的默认选择,但是取决于样品本身。
4、检测器
有足够的生色团,
UV检测器是首选。当不能被UV检测时,蒸发光散射或者其他检测器也适用。
5、分离条件的选择
对于大多数样品,可根据连续
3个实验以一个系统化、反复测试的方法来进行。
首先:改变
B值,实现合适的保留范围(1≤k≤10).
其次:应对不同的分离条件来贺寿的选择性(
α)和分离度。首先改变的是%B(
±10%B)和温度。
再次:改变色谱柱就:长度、颗粒大小、流速等。
或者搜索文献。
6、潜在问题的评估、识别和解决之道
在
HPLC的建立和随后的应用中,峰形,保留试剂的飘逸会马上显示,有些问题可以事先预测减少发生可能:
A
:极性的的样品无保留
B:被忽视的色谱峰
C:不耐受的分离条件
D;设备之间的差异
解决办法:
A极性样品的无保留:可以使用正相色谱、HILIC、离子对色谱法、离子交换色谱法等
B:色谱峰忽视:1.检测器灵敏度不够
2. 色谱程序不能分离相邻的两峰,峰重叠
使用
MS检测的手段或者采用正交分离的法。被忽视的色谱峰可能会在日常操作中显现出来,需要进一步分离。
C:不耐受的分离条件:因为分离条件微小的不明显的变化造成分离度的损失。(如流动相ph)通常需要改变分离条件来提高耐用性。
D:可能存在的设备差异。在同一厂家不同仪器和不同厂家的仪器之间,由于不同系统的死时间存在差异会使方法特别是进行梯度洗脱的时候造成分离差异。需要在不同仪器之间进行实验,必要时可以加入梯度延时减少差异。
7、方法的确认和系统适应标准的确定
在正式方法确认之前可以展开
”
预验证
”(精确度、准确度、线性、分离完整批次样品的能力),防止正式实验时出现问题。
本文是药群论坛群友郭巧灵的读书笔记,观点仅代表作者本人,如需转载,需经过药群论坛和作者授权!
古人说,书中自有黄金屋,
书中自有颜如玉
,
外国人说,书籍是人类进步的阶梯,读书,就是灵魂升华的过程,心得,就是知识融合的舍利,来吧,朋友,走进书籍的世界,来吧,朋友,谈谈读书的心得,分享你的读书故事给我,
投稿发微信号dieerfeiya,QQ号404760205
-----------------------------------------------------------
药群论坛的QQ群
药品研发&注册总群 142331258;
药品研发&注册高级群 102700857;
药品GMP&生产总群 219048542;
药品质量检验总群 300939182;
生物制品注册研发总群 487476133;
药品临床研究总群 322401932;
药品临床前研究总群 149677623;
医疗器械研发注册群450482666;
药包材&辅料注册主群 522526281;
药品一致性评价总群 564089386;
保健食品研发注册群 542880379;










