为了更好的阅读体验,请点击下方阅读原文在简书中查看
本教程来自于李继存老师博客:http://jerkwin.coding.me/GMX/GMXtut-1/#%E6%A6%82%E8%BF%B0
特此说明
今天带和大家介绍一下gromacs最简单的教程,这个教程李老师已经组织翻译了一遍,翻译的质量非常高,我仍然炒一遍剩饭主要是想准备把自己所学的温故知新一遍,为了更加快速的进行模拟,我们将两个教程的蛋白进行了替换,即将載脂蛋白替换了溶菌酶,实际上是一样的。
首先看一下GROMACS做MD的一般流程
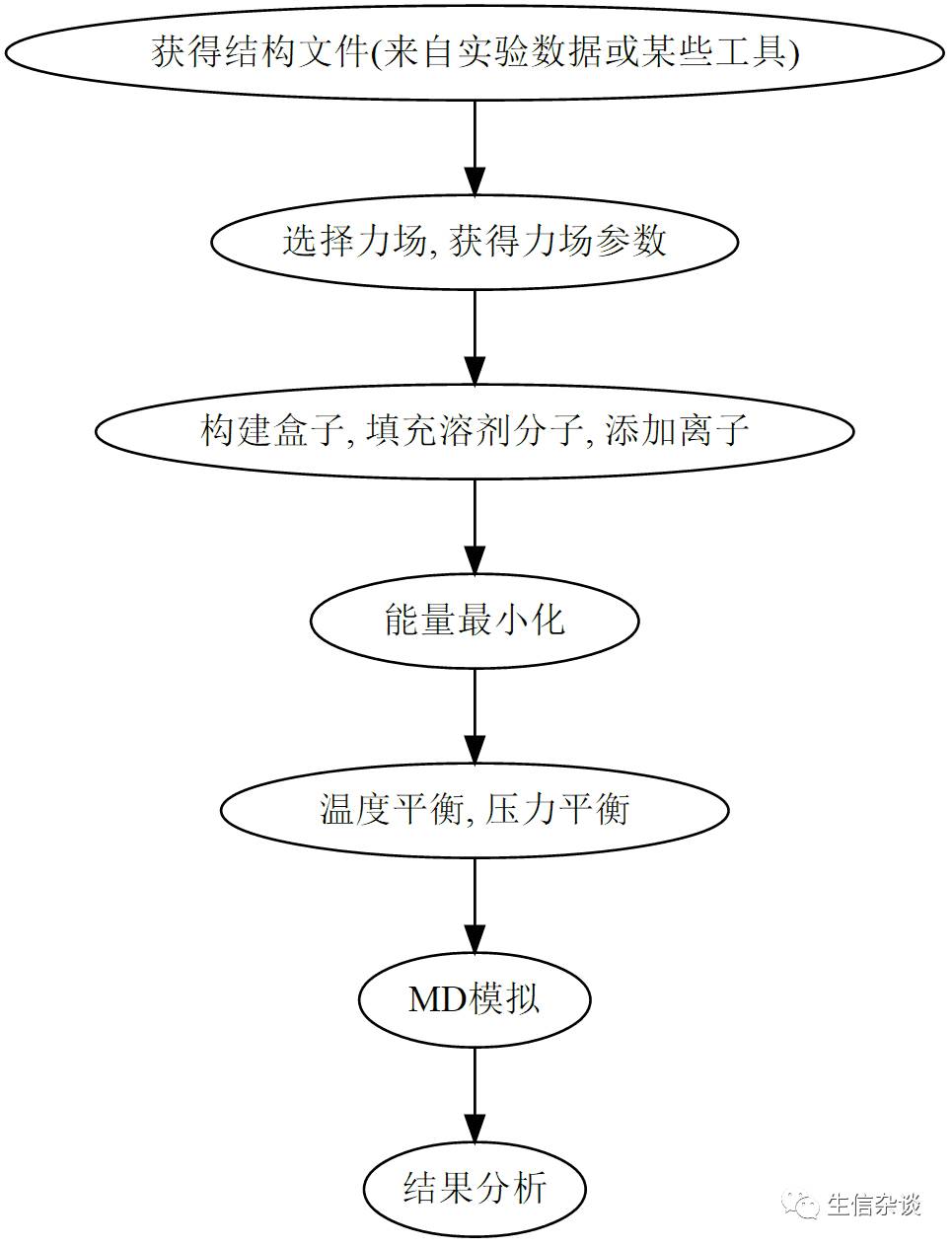
下载PDB文件
首先我们在PDB数据库下载小肽的PDB文件,注意我采用的为jupyter notebook编写,若在终端运行的话无需加out=!{}
例如:out=!{“ls”},在终端运行只需要为ls即可
out=!{"wget http://www.rcsb.org/pdb/files/1ODQ.pdb"}
生成蛋白拓扑(Topology文件)
在用GROMACS的程序对pdb文件进行处理前, 要做许多检查, 以保证pdb文件的完整性. 根据pdb文件的不同, 要进行不同的处理. 需要处理的方面包括但不限于:
-氢原子
-C端氧原子
-结晶水
-缺失原子/残基/侧链
-二硫键
-带电残基
请在进行下一步之前仔细简单是否有错误,如教程蜘蛛肽需要进行末尾蛋白的补全,如删除蛋白中本身含有的结晶水等等工作。
在本例中你需要从20个模型中获取单一模型,可以使用例如
pymol
进行操作。
下一步需要注意的是我们需要
-ignh
进行删除pdb中的氢,然后gmx进行自动加氢。我们将其修改为fws.pdb
!gmx pdb2gmx -f fws.pdb -ignh -o 1ODQ_processed.gro -water spce
····
Select the Force Field:
From '/usr/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
力场包含了将要写入到拓扑文件中的信息. 这个选择非常重要! 你必须仔细了解每个力场, 并决定哪个力场最适用于你的体系. 在本教程中, 我们选gromos联合原子力场, 因此在命令提示行中输入
14
, 然后回车.由于在jupyter中shell交互不是很方便,所以我们采用的是在尾部
<
的方式进行交互,下面的情况类似,不再申明,
即大家在实际操作中在shell终端输入前面的命令,后面选择的选项与后面命令类似
。
out=!{"gmx pdb2gmx -f fws.pdb -ignh -o fws_processed.gro -water spce <}
定义单位盒子并填充溶剂
教程中是需要模拟一个简单的水溶液分子,定义模拟用的盒子并添加溶剂分为两步完成:
1.使用
editconf
模块定义盒子的尺寸
2.使用
solvate
模块想盒子中填充水
首先我们来定义盒子,本教程用的正方形盒子,其实盒子种类很多,一般使用菱形十二面体晶胞,其可以节省许多水分子,从而节省计算量。
-c
表示分子在盒子的中心部分。
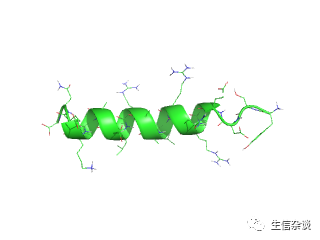
out=!{"gmx editconf -f fws_processed.gro -o fws_newbox.gro -c -d 1.0 -bt cubic"}
我们可以在vmd或者pymol中查看分子的情况,在用shell交互中下面块的内容不需要输入,转而使用
vmd fws_newbox.gro
或者
pymol fws_newbox.gro
进行查看
from IPythonTweaks import *
pymolPlotStructure("fws_newbox.gro")
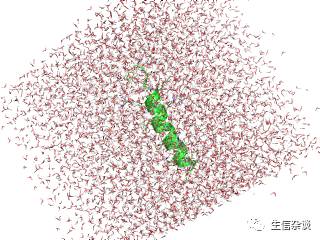
之后我们进行溶剂分子的增加
out=!{"gmx solvate -cp fws_newbox.gro -cs spc216.gro -o fws_solv.gro -p topol.top"}
同样我们也可以进行可视化查看
pymolPlotStructure("fws_solv.gro")
增加离子
现在我们已经有了一个带电荷的溶液体系,
其实在pdb2gmx
程序的输出文件会显示带了多少电荷,如果没有查看到我们可以查看一下
topol.top
文件中
[ atoms ]
指令的的最后一行, 它应该含有
qtot 0
这一信息. 由于生命体系中不存在净电荷, 所以我们必须往我们体系中添加离子, 以保证总电荷为零.虽然电荷为0,我们还是增加一下0个离子,作为演示的作用。
首先我们创建一个ions.mdp的文件,并输入如下内容:
; ions.mdp - used as input into grompp to generate ions.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.0 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions (yes/no)
file=open("ions.mdp","w")
file.write("""
; ions.mdp - used as input into grompp to generate ions.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.0 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions (yes/no)
""")
file.close()
使用如下命令来产生
.tpr
文件
out=!{"gmx grompp -f ions.mdp -c fws_solv.gro -p topol.top -o ions.tpr"}
out=!{"gmx genion -s ions.tpr -o fws_solv_ions.gro -p topol.top -pname NA -nname CL -nn 0 <}
out
···
'GROMACS: gmx genion, VERSION 5.1.2',
'Executable: /usr/bin/gmx',
'Data prefix: /usr',
'Command line:',
' gmx genion -s ions.tpr -o fws_solv_ions.gro -p topol.top -pname NA -nname CL -nn 0',
'',
'Reading file ions.tpr, VERSION 5.1.2 (single precision)',
'Reading file ions.tpr, VERSION 5.1.2 (single precision)',
'No ions to add, will just copy input configuration.',
'',
'gcq#418: "A curious aspect of the theory of evolution is that everybody thinks he understands it." (Jacques Monod)',
'']
能量最小化
现在, 我们已经添加了溶剂分子和离子, 得到了一个电中性的体系. 在开始动力学模拟之前, 我们必须保证体系的结构正常, 原子之间的距离不会过近, 几何构型合理. 对结构进行弛豫可以达到这些要求, 这个过程称为能量最小化(EM, energy minimization).
能量最小化过程与添加离子过程差不多. 我们要再次使用
grompp
将结构, 拓扑和模拟参数写入一个二进制的输入文件中(.tpr), 但这次我们不需要将
.tpr
文件传递给
genion
, 而是使用GROMACS MD引擎的
mdrun
模块来进行能量最小化.
能量最小化的
mdp
文件参数与解释如下:










