后
GWAS
时代人们最关心的问题是如何解释显著遗传位点的生物学功能。一般来讲,由
GWAS
找到的显著位点大部分落在不编码蛋白的基因组区域,然而我们对这些区域的了解目前还十分有限。随着
ENCODE, FANTOM
等数据库的逐渐完善,基因组中的非编码区段正在被各种新的组学技术揭开神秘面纱。具有某些特定功能的基因组非编码区段是否与
GWAS
易感位点在位置上具有显著的交互关系就显得尤为重要。
为了帮助研究以上科学问题,我给各位读者介绍一款不错的工具——
Variant Set Enrichment (VSE).
首先,这是一个非常有名的
R
包,曾在《
Nature Genetics
》被多次引用。其核心算法是比较真实的遗传位点集合是否比随机匹配的模拟集合更加富集与某一特定的功能基因组区段上。
下面我就带领大家利用示例数据学习使用
VSE
。
首先需要说明的是
GWAS
报道的显著位点以及与其有连锁不平衡的位点都有可能发挥生物学功能,因此我们需要将
tag SNP
以及其
LD SNP
一起纳入分析。
VSE
支持直接从
rAggr
网站(
http://raggr.usc.edu/
)上获取的具有连锁关系的
SNP
集合。一旦完成以上工作,数据导入就很简单了。
示例代码如下:
library("VSE")
bca.ld
bca.avs
avs.size
这一步中,
VSE
要根据第一步得到的真实的位点信息去基因组中随机产生匹配的模拟位点。只用一行代码就可以轻松搞定。
###该步骤有两个参数,其中bgSzie表示随机模拟的次数,mc.cores表示用到的计算机节点数,Windows下只能是1
还是经典的
bed
文件格式即可。
VSE
自带了
5
个来自
MCF7
细胞系的组蛋白
Chip-seq
的数据,下载地址为(
www.hansenhelab.org/VSE/sample_regions/
)。读者也可以通过自带函数“
loadSampleRegions
”下载。
# Downloading sample regions
sampleSheet_path
# Loading sample sheet
samples
在进行统计分析之前,
VSE
可以给出一个近似热图的交互矩阵(如图
1
)。不禁让人窃喜:又能在文章中多放一个图了。
图
1
:遗传位点与基因组区段的交互热图

VSE
需要
SNP
集合,模拟集合以及基因组坐标三个输入来进行富集分析。一行代码即可实现:
在展示
P
值之前,
VSE
还提供
QQ plot
对
null distribution
的正态性进行检验。只有符合正态分布的结果才可行。
par.original
par(mfrow = c(ceiling(length(samples$Peaks)/3), 3), mai = c(1, 1, 0.5, 0.1))
VSEqq(bca.vse)
par(par.original)
如图
2
,富集分析的正态性进行检验结果
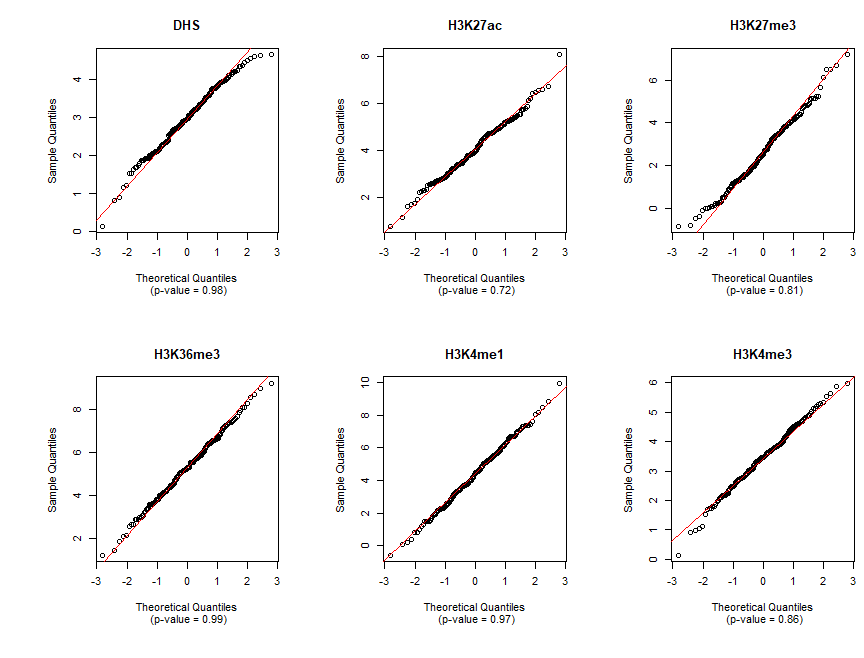
最后,我们可以放心大胆地展示结果了。
VSE
提供图和表两种形式的展示结果的方式。
(
i
)表
结果如下:

(
ii
)图
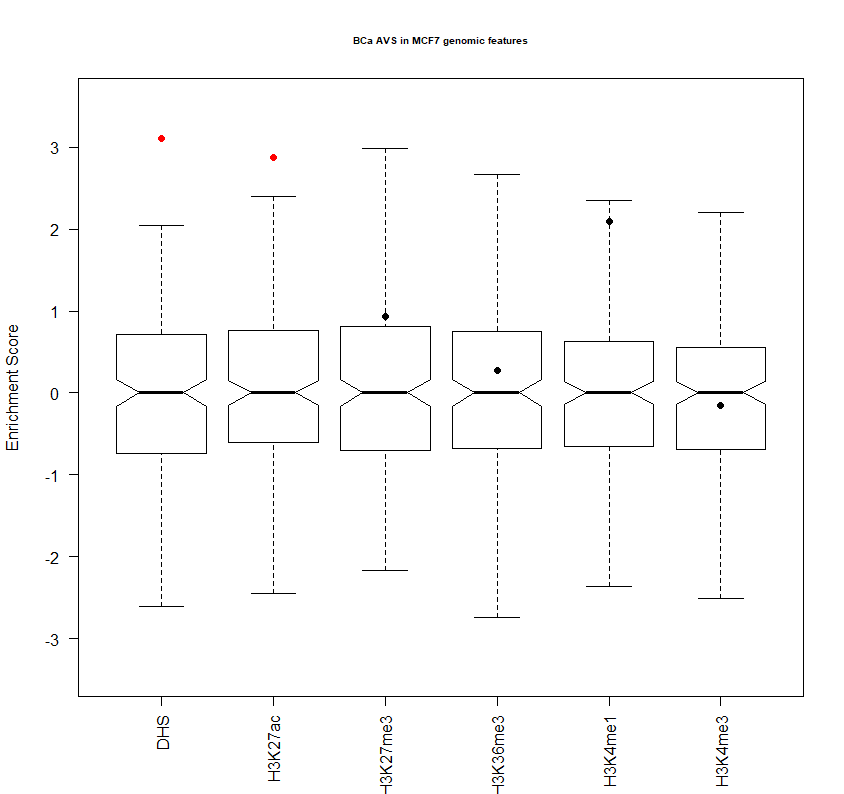
VSEplot(bca.vse, las = 2, pch = 20, cex = 1, cex.main = 0.6, padj = 0.05, main = "BCa AVS in MCF7 genomic features")
如图
3
,红色的点对应的区域既是遗传位点显著富集的区域(
Bonferroni adjusted P-value < 0.01
)
图
3
:最终富集结果图
(1)VSE对tag SNP的数量非常敏感,低于15会导致不准确的结果。
(2)确保使用0.8作为LD的阈值。
(3)用户需尽量选择可靠来源的功能基因组数据。
(4)随机次数决定结果的显著程度。随机次数越大,分布的正态性越好,结果越可信。
本文为生信草堂原创,欢迎个人转发分享,其它媒体或网站如需转载,请在正文前注明转自生信草堂并联系bioinformatics88
又见数据挖掘|遗传与调控数据揭示肺癌亚型之间的若即若离
功能基因组学研究利器——Hi-C
如何从ENCODE数据库中快速获取组蛋白chip-Seq的可视化数据
功能基因组学研究利器——ChIA-PET





