慢病毒载体(Lentivirus)
作为一个常规的病毒实验载体已经熟用于我们的日常实验中,但是其是否可以安全的用于临床治疗一直饱受争议。继CAR-T细胞治疗后,近日,又一轰动性的研究成果展示了慢病毒载体的无限可能。
2018年4月19日出版的
《新英格兰医学杂志》
(NEJM,影响因子:72)发表论著《输血依赖型β-地中海贫血患者的基因治疗》(Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia)。研究者,
利
用了慢病毒介导的基因治疗方案——LentiGlobinBB305对β地中海贫血患者进行了治疗,并获得了显著疗效
,
同时显示,慢病毒载体的基因疗法是安全的。
该新型基因疗法可使输血依赖型β地中海贫血(TDT)患者免于或减少长期输血。

|
背景资料
地中海贫血(
Thalassemia)于1925年由Cooley和Lee首先描述,最早发现于地中海区域,当时称为地中海贫血,国外亦称海洋性贫血。这是一类由于常染色体遗传性缺陷,引起珠蛋白链合成障碍,使一种或几种珠蛋白数量不足或完全缺乏,因而红细胞易被溶解破坏的溶血性贫血。
珠蛋白链
的分子结构及合成是由基因决定的。γ、δ、ε和β珠蛋白基因组成“β基因族”,ζ和α珠蛋白组成“α基因族”。正常人自父母双方各继承2个α珠蛋白基因(αα/αα)合成足够的α珠蛋白链;自父母双方各继承1个β珠蛋白基因合成足够的β珠蛋白链。由于珠蛋白基因的缺失或点突变,肽链合成障碍导致发病。地中海贫血分为α型、β型、δβ型和δ型4种,其中以
β和α
地中海贫血
较为常见
。
1. β珠蛋白生成障碍性
贫血
(β
地中海贫血
)
β珠蛋白生成障碍性贫血(简称β地贫)的发生的分子病理相当复杂,已知有100种以上的β基因突变,主要是由于基因的点突变,少数为基因缺失。
2. α珠蛋白生成障碍性
贫血
(α
地中海贫血
)
大多数α珠蛋白生成障碍性贫血(地中海贫血)(简称α地贫)是由于α珠蛋白基因的缺失所致,少数由基因点突变造成。白基因的缺失所致,少数由基因点突变造成。
目前,有效的治疗手段有异源性造血细胞移植、长期规律性红细胞注射和祛铁治疗,但是有排斥风险,并发症,和其他毒性效应。因此基因治疗方案正在被评估作为一种新的方案。(来源于:百度百科
|
治疗流程
-
在美国对病人给药filgrastim 和 plerixafor处理,收集病人造血干细胞与原代细胞;
-
富集CD34+细胞,并对细胞进行BB305慢病毒载体转导处理,得到的药物产品命名为LentiGlobin,低温储藏;
-
对病人进行连续4天的白消安( busulfan )骨髓抑制处理,(注射剂量依据病人血清白消安药代动力学分析结果来调整);
-
72小时监察,确定病人达到医学认定的稳定期(连续3天病人每立方毫米血样中有至少500个中性粒细胞);
-
在HGB-204临床实验中,β0/β0基因型病人注射剂量中位值为11.0(6.1-18.1)百万CD34+细胞,其他基因型注射7.2(5.2-13.0)百万CD34+细胞;
-
在HGB-205临床实验中,1位IVS1-110突变基因型病人注射8.8百万CD34+细胞,其余3位βE/β0基因型病人注射剂量中位值为12.0(8.9-13.6)百万CD34+细胞
(注射量见下表);
-
综合分析各项检测指标并进行安全性评价。

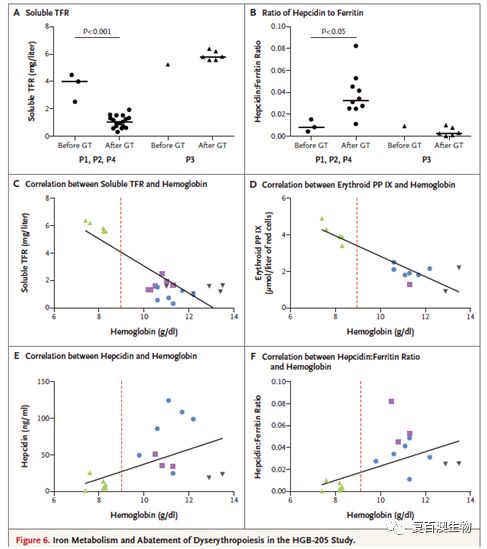
经过lentiGlobin药物静脉注射治疗后,植入性中性粒细胞和血小板在中位值17天及30天出现,期间未有流血并发症出现。载体在PBMCs中的平均拷贝数如下图所示,在HGB-204的每二倍体基因组中的拷贝数为0.3,在HGB-205的拷贝数为2.0。且在PBMCs中的拷贝数与载体在药物中的拷贝数成正相关。
病人血液中HBAT87Q蛋白水平提高,对红细胞注射依赖量的减少73%。
 在22位病人中,13位为非β0/β0基因型,除一人外(其注射药物中载体拷贝数为0.3,注射后体内为0.1,拷贝数太少)其余均停止了红细胞注射治疗,HbAT87Q含量中位值是6.0g每分升,血红蛋白含量为11.2。9位β0/β0基因型病人,与两年前未接受治疗相比,对红细胞注射的需求量减少73%。
在两项研究中,血液中HbAT87Q表达水平与血液中载体拷贝数相关,与年龄、基因型、脾切除状态无关
。
在22位病人中,13位为非β0/β0基因型,除一人外(其注射药物中载体拷贝数为0.3,注射后体内为0.1,拷贝数太少)其余均停止了红细胞注射治疗,HbAT87Q含量中位值是6.0g每分升,血红蛋白含量为11.2。9位β0/β0基因型病人,与两年前未接受治疗相比,对红细胞注射的需求量减少73%。
在两项研究中,血液中HbAT87Q表达水平与血液中载体拷贝数相关,与年龄、基因型、脾切除状态无关
。
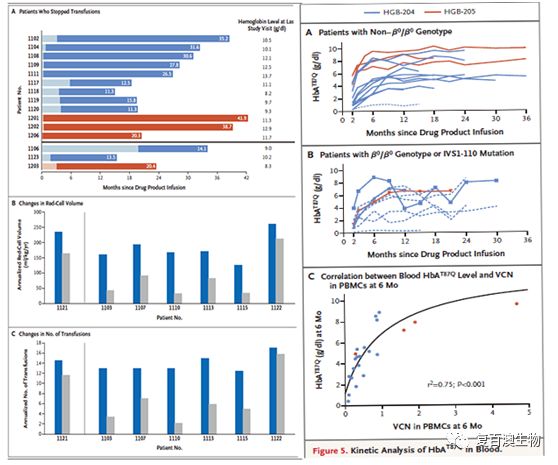
总体上病人血红蛋白趋于稳定,溶血现象趋于缓解。红系造血异常现象在3位βE/β0基因型中被消除,在IVS1-110基因突变型病人中无效造血现象的生物标记物也未被发现。