
第一作者:Yuchen Han,Lei Zhao
通讯作者:徐坤,林运祥
通讯单位:安徽大学化学化工学院,物质科学与信息技术研究院
论文DOI:10.1002/adfm.202407060
本研究采用焦耳热快速加热和传统管式炉煅烧两种方法成功制备了两种不同晶相结构的碳化钼,即立方相α-MoC
1-x
和六方相β-Mo
2
C。随后,通过浸渍还原方法,在不同晶相结构的碳化钼基底上负载了少量的Ir团簇,制备出Ir/α-MoC
1-x
和Ir/β-Mo
2
C两种模型催化剂。研究结果显示,锚定在不同类型碳化钼基底上的Ir团簇,通过可调控的电子相互作用,在碱性HOR中展现出不同的性能。值得注意的是,具有较强电子相互作用的Ir/α-MoC
1-x
的质量活性约为Ir/β-Mo
2
C的两倍。实验和理论计算均证实,Ir/
α
-MoC
1-x
可能通过界面的Ir-Mo键实现了强的金属-基底相互作用,显著调制了电子结构并优化了催化剂的H和OH结合能,从而促进了碱性HOR的反应动力学过程。这项研究工作强调了可通过调控载体的晶相结构有效调节金属负载型催化剂的局部电子结构,进而提高催化剂的本征催化活性。
开发低贵金属含量的碱性HOR催化剂对推动阴离子交换膜燃料电池的发展具有重要意义。构建原子级铂族金属负载型电催化剂是发展高性价比碱性HOR电催化剂的常用策略。过渡金属碳化物(TMCs)
因其结构组成可调、类铂的电子结构、稳定性优异等优点被广泛认为是负载型电催化剂最有潜力的载体之一。然而,基于TMCs的金属负载型碱性HOR催化剂仍处于初步阶段。如何实现原子级贵金属与碳化物基底之间电子相互作用的精细调控进而实现更高效的电催化活性是里面的关键科学问题
1. 制备了两种不同晶相结构的碳化钼负载型Ir/α-MoC
1-x
和Ir/β-Mo
2
C模型催化剂,呈现出不同的催化活性。其中,Ir团簇负载在立方相α-MoC
1-x
的质量活性约为其负载在六方相β-Mo
2
C的两倍。
2. 结合系列实验和理论计算证实Ir/α-MoC
1-x
通过界面Ir-Mo键的强金属-基底相互作用,有效调节了催化剂的局部电子构型并优化了反应中间体H*和OH*的结合能,从而获得优异的碱性HOR催化性能。
图1. Ir/α-MoC
1-x
的形貌及微观结构表征。(a) XRD图谱。(b, c) 不同尺度的TEM图像。(d) HAADF-STEM图像。(e, f) HAADF-STEM图像中绿色虚线框和橙色圆圈标注对应的晶格结构。(g-j) HADDF-STEM图像和相应的EDX元素分布图。
从透射电镜和高分辨电镜上可以看出,合成的α-MoC
1-x
主要以颗粒的形式嵌入花状形态的碳骨架结构中。Ir/α-MoC
1-x
的高角度环形暗场扫描TEM (HAADF-STEM)图像表明Ir团簇主要分布在α-MoC
1-x
表面。同时,Ir/α-MoC
1-x
对应的元素分布图结果表明C、Mo和Ir三种元素是均匀分布的。
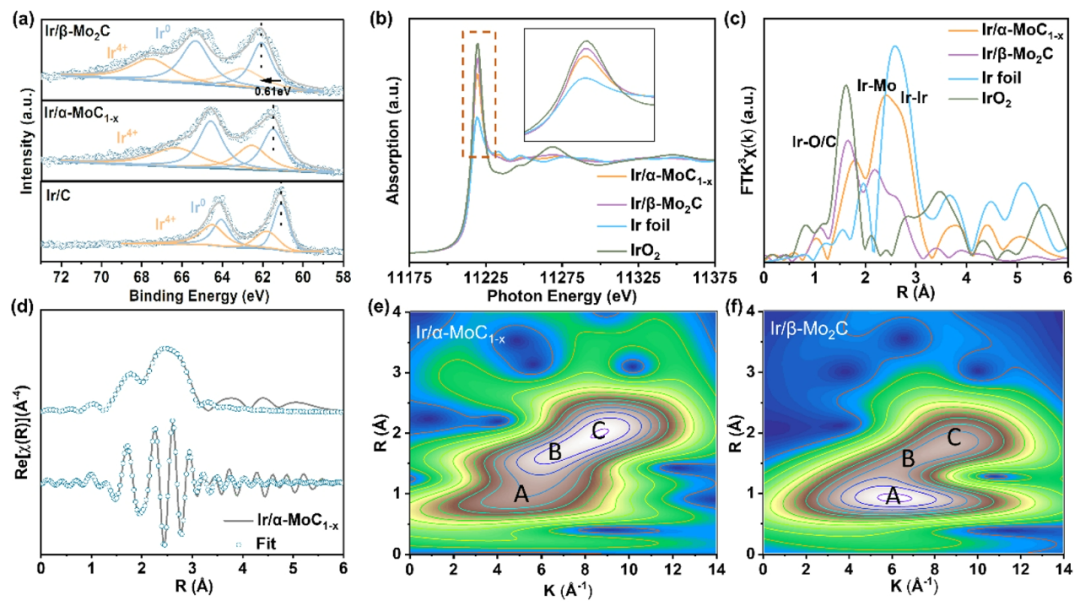
图2. Ir/α-MoC
1−x
和对比样品的电子结构表征。(a) Ir 4f XPS光谱。(b) Ir L
3
边XANES光谱。(c) Ir L
3
边FT-EXAFS曲线。(d) Ir/α-MoC
1-x
在R空间的FT k
2
加权谱和拟合曲线。(e,f) Ir/α-MoC
1-x
和Ir/β-Mo
2
C的WT-EXAFS曲线。
与Ir/β-Mo
2
C相比,Ir/α-MoC
1-x
中Ir 4f光谱显示出轻微的负偏移,这表明α-MoC
1-x
可以为Ir团簇提供更多电子。此外,Ir L
3
边的X射线吸收近边结构(XANES)谱图显示出不同特征。Ir/α-MoC
1-x
的白线峰强度明显低于Ir/β-Mo
2
C和IrO
2
,这证实了Ir/α-MoC
1-x
中Ir的价态较低,同时也说明了Ir团簇与α-MoC
1-x
和β-Mo
2
C之间存在着不同程度的电荷转移。上述结果表明,α-MoC
1-x
可能提供了一个特殊的电子转移通道,使电子能够更多的从基底转移到Ir团簇上。傅立叶变换扩展X射线吸收精细结构(FT-EXAFS)光谱分析表明,两处散射峰位于1.64 Å和1.77 Å,分别对应Ir团簇与碳化钼基底之间所形成的化学键和表面氧化作用所形成的Ir-C/O键。在R空间光谱中,可以明显观察到Ir-Mo键的存在。通过EXAFS拟合,对Ir的局部配位信息进行了表征。与β-Mo
2
C相比,Ir/α-MoC
1-x
表现出较低的Ir-C配位数和较高的Ir-Mo配位数,这表明了Ir团簇与α-MoC
1-x
之间由于Ir-Mo键的存在进而可能产生了更强的相互作用。
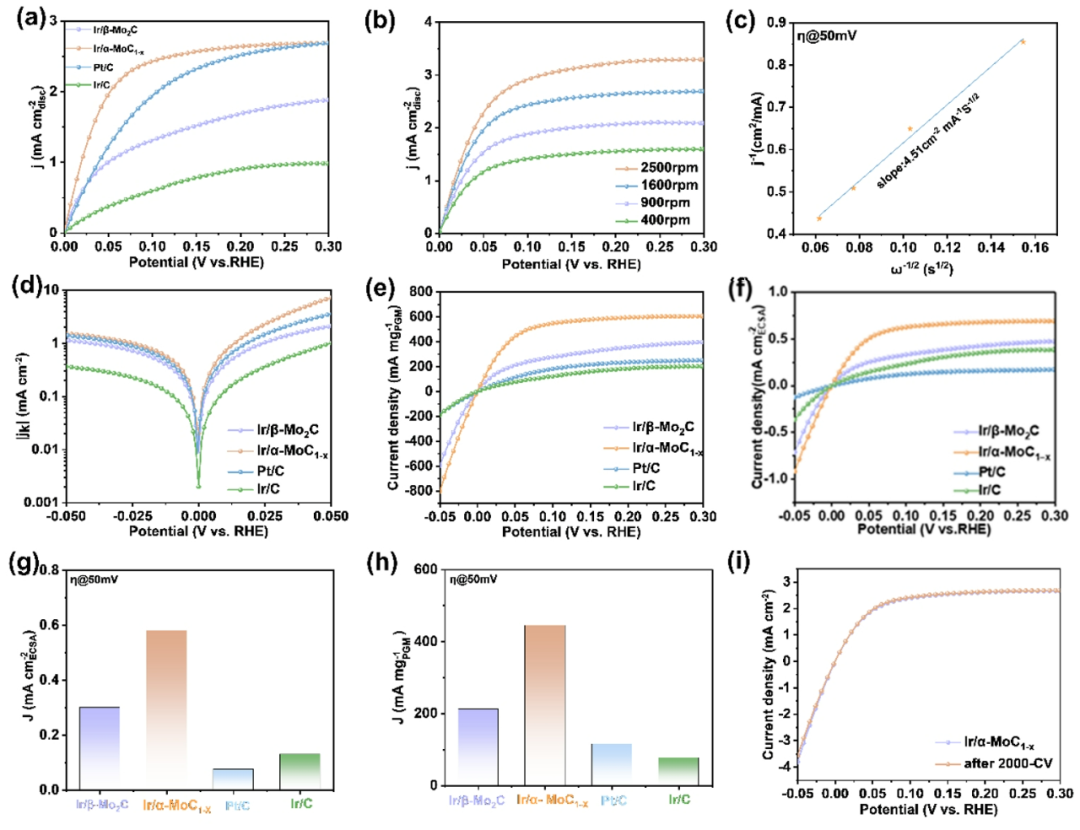
图3. 碱性HOR的电化学性能。(a) 不同催化剂在H
2
饱和的0.1 M KOH电解质中的HOR极化曲线。(b) 不同旋转速率下Ir/α-MoC
1-x
的极化曲线。(c)微极化区域的线性拟合。(d) HOR动力学电流密度(j
k
)。(e) 样品的质量归一化电流密度。(f) 样品的ECSA归一化电流密度。(g) 过电位为50
mV时样品的ECSA归一化电流密度。(h)过电位为50 mV时样品的质量归一化电流密度。(i) Ir/α-MoC
1-x
经过2000次循环后的稳定性测试。
Ir/α-MoC
1-x
在50 mV时的电流密度为1.96 mA cm
-2
,明显高于其他对比样品。Ir/α-MoC
1-x
在50 mV时的电化学面积归一化电流密度分别是Ir/β-Mo
2
C、Pt/C和Ir/C的1.96倍、6.37倍和3.42倍。同时,Ir/α-MoC
1-x
在50 mV下的质量活性为445 mA mg
-1
Ir,分别是Ir/β-Mo
2
C、Pt/C和Ir/C的2.11倍、3.91倍和5.78倍。经过2000次循环CV测试后,催化剂的活性下降可以忽略不计,证明了Ir/α-MoC
1-x
具有较好的稳定性。
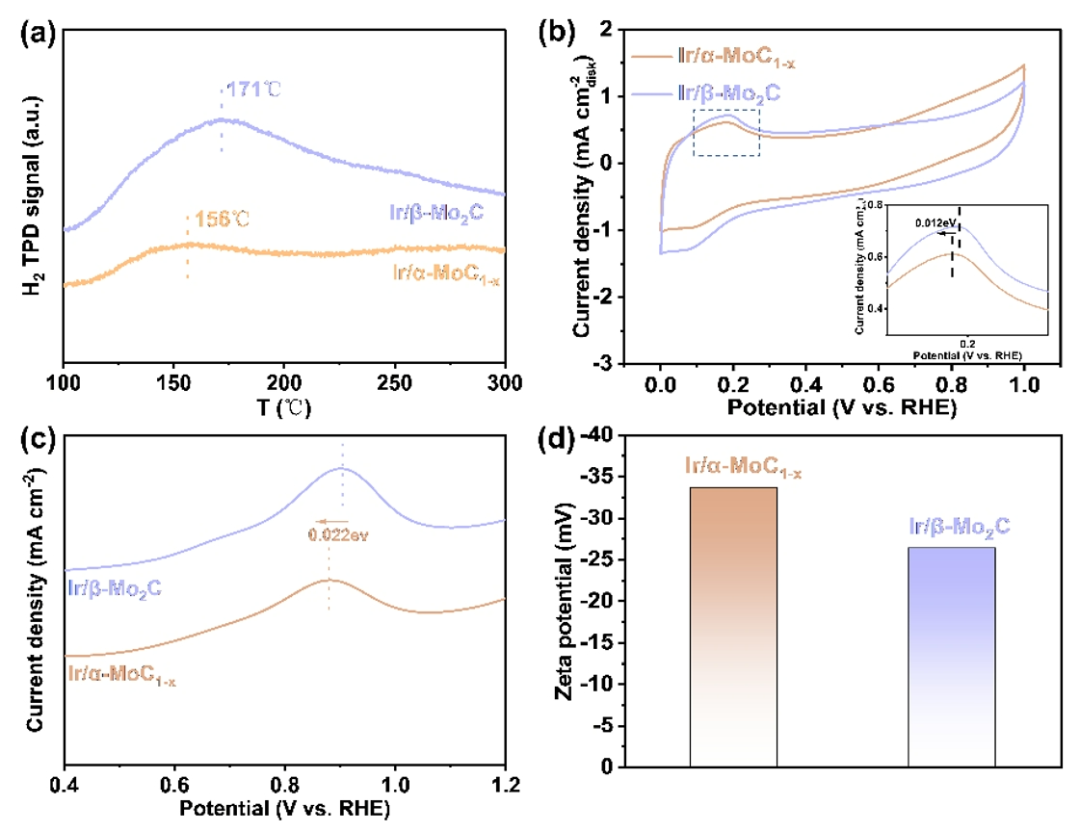
图4. 机理研究。(a) Ir/α-MoC
1-x
和Ir/β-Mo
2
C的H
2
-TPD测试。(b) H
UPD
。(c) CO溶出曲线测试。(d) Zeta电位。
HBE和OHBE是碱性HOR催化剂活性的两个关键描述符。通过H
2
程序升温脱附(H
2
-TPD)和欠电位沉积氢(H
UPD
)表明Ir/α-MoC
1-x
的HBE较弱。OHBE主要通过CO溶出实验和Zeta电位测试两种途径来进行测定,结果表明,Ir/α-MoC
1-x
具有更强的OHBE。
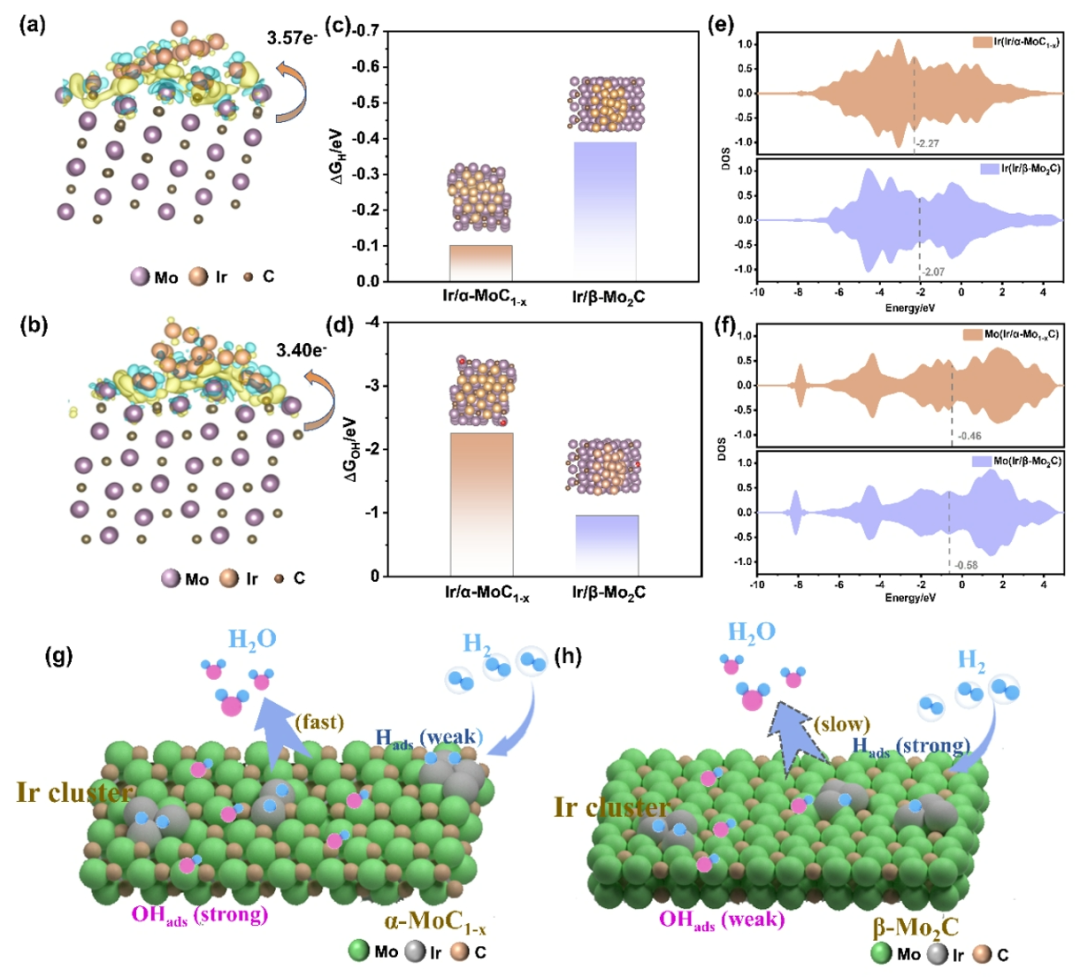
图5. 密度泛函理论计算。(a, b) Ir/α-MoC
1-x
和Ir/β-Mo
2
C的差分电荷密度。(c, d) H和OH在Ir/β-Mo
2
C和Ir/α-MoC
1-x
表面的吸附位点以及相应的吸附结合能ΔE
H
和ΔE
OH
。(e, f) 不同模型吸附*H和*OH的态密度(DOS)图。(g, h) Ir/α-MoC
1-x
和Ir/β-Mo
2
C在碱性HOR反应过程中催化机理的示意图。
通过理论计算对Ir/α-MoC
1-x
和Ir/β-Mo
2
C之间碱性HOR性能差异进行探讨。Bader电荷分析表明Ir团簇与立方相α-MoC
1-x
基底之间发生的电荷极化更强,电荷积累量更高,这可能是由于Ir-Mo键的存在引起的。进一步,计算了催化表面的HBE和OHBE。与H结合相关的Ir活性位点的d带中心和与OH结合相关的Mo活性位点的d带中心等理论计算结果均表明H 在Ir/α-MoC
1-x
的吸附较弱,而OH的吸附较强,与实验结果相符合。因此,相较于Ir/β-Mo
2
C,弱化的HBE以及增强的OHBE可能是Ir/
α
-MoC
1-x
在碱性HOR中表现出加快的反应动力学原因。
Ir/
α
-MoC
1-x
由于Ir团簇与立方相MoC
1-x
较强的电子相互作用使得其呈现出优异的HOR电催化活性。实验结果与理论计算表明Ir/
α
-MoC
1-x
中Ir- Mo间隙键的存在可能会增强Ir团簇与碳化钼基底之间的相互作用,进而优化催化剂的电子结构得到促进HOR反应动力学。这项工作有望对负载型原子尺度贵金属基电催化剂设计提供新的思路。
欢迎关注我们,订阅更多最新消息









