【作者】知行,格罗宁根大学 代谢组学博士在读
【责编】Ryan
第一步是蛋白酶切(Digestion) 成肽段
其中最常用的是胰蛋白酶,它在精氨酸(Arg)和赖氨酸(Lys)位点将蛋白酶切。酶切后的大部分肽段会按照胰蛋白酶的酶切规则变成“胰蛋白酶酶切肽段”。将蛋白质酶解后产生复杂的肽段混合物会非常复杂,因为胰蛋白酶酶切后会平均产生50条肽段。因此在蛋白质酶解之前一般采用SDS-PAGE进行预分离(Seperation),以将所有的蛋白质组分分离成子蛋白质组分来减少样品复杂性。蛋白质酶解后通常会进行肽段选择,以挑选特定属性肽段(如糖基化肽段、磷酸化肽段等等)。输出的肽段样品使用反向色谱层析法进一步分离。
第二步是将酶切肽段送入质谱仪分析
肽段从反相色谱柱出来后被离子化、气态化,之后特定的离子进入串联质谱仪(MS/MS)以生成碎片离子质谱(MS/MS spectra)。
数据获取过程如下:质谱仪扫描所有肽段离子并记录一级质谱的质荷比(m/z)和丰度。之后经选择的肽段离子(也称为母离子precursor)在质谱仪的碰撞池中裂解成更小的碎片离子。获得的二级质谱(MS/MS)是由母离子生成的所有碎片离子的质荷比和丰度。根据MS/MS质谱仪的裂解规律可以识别产生碎片离子的肽段的氨基酸序列。碎片离子的生成通常采用碰撞诱导解离技术(CID)、电子传递解离(ETD)和高能碰撞解离(HCD)。有一些更高级的质谱仪可以将碎片离子进一步碎裂,从而生成三级质谱。质谱分析器的质量精确度和分辨率对质谱信息有一个显著的影响,从而影响接下来的肽段鉴定。质谱仪的精确度最低可以达到几百万分之一(ppm),比如LTQ-Orbitrap这种高质量精确度的仪器。而对于低质量精确度的仪器,其精确度会高达500 ppm。即使是采用高质量精确度的仪器,要获得真正的高质量精确度,还需要经过优化仪器设置、控制室内温度、计算校准等步骤。质谱仪的质量分辨率意味着可以精确确定肽段离子电荷状态的能力,高质量分辨率可以在一个较窄的质荷比范围内分辨MS/MS质谱数据,从而排除无关数据的影响。
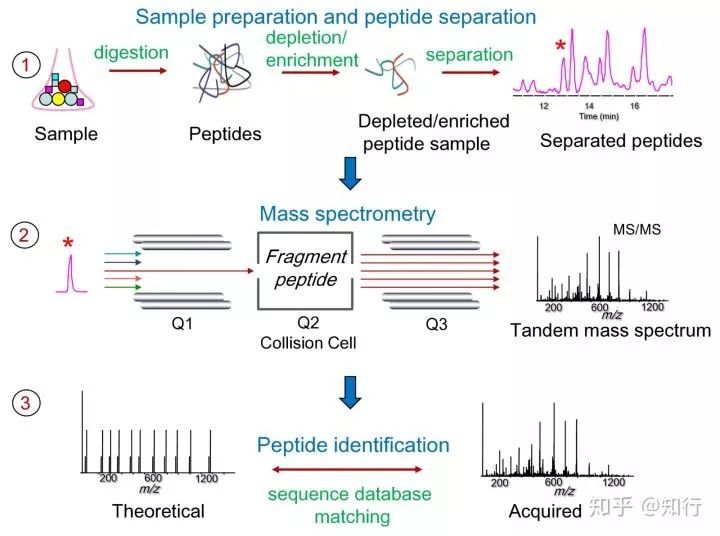
第三步是进行肽段鉴定(Peptide identification)
分析的关键是根据MS/MS质谱数据鉴定出对应的肽段。而肽段鉴定策略可大致分为以下四类:序列数据库搜索方法(database search approach)是通过比较实验谱图和根据蛋白质序列数据库理论酶切形成的理论谱图来鉴定肽段;谱图库搜索方法(spectral library searching)是通过直接比较实验谱图和理论谱图来鉴定肽段;而从头测序方法(de novo sequencing approach)直接从质谱数据推测肽段而无需数据库的帮助;肽序列标签法是数据库搜索方法和从头测序方法的整合,它先通过从头测序方法鉴定3-5个残基长度的短序列标签,之后再通过数据库搜索方法鉴定整个肽段。
本文仅介绍主流的序列数据库搜索方法(Sequence database search approach)。如下图所示:
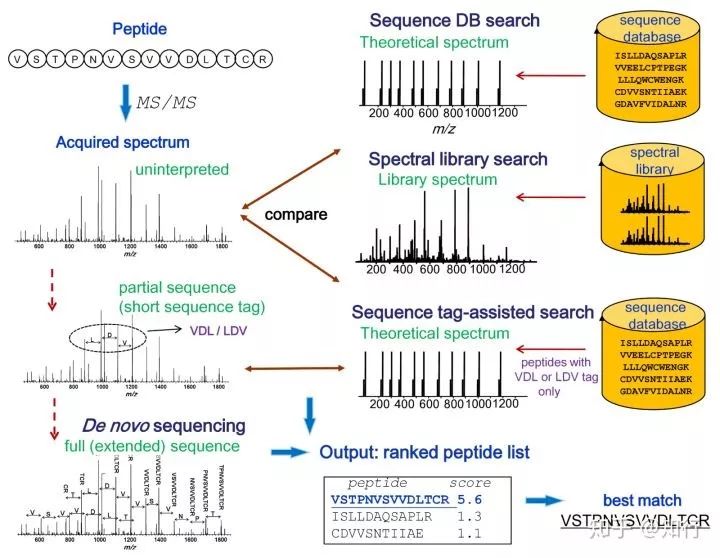
因为采用胰蛋白酶酶切,所以酶切后的肽段以氨基酸R结尾。该酶切肽段送入串联质谱分析后获得质谱(Acquired spectrum)。序列数据库搜索软件以实验MS/MS质谱数据与计算机模拟的理论酶切质谱数据(Theoretical spectrum)相比较。通过计算机模拟理论酶切并设置过滤参数从而得到目标序列。最重要的参数包括母离子质量误差容限、酶切类型、固定修饰、可变修饰。其他的参数包括碎片离子类型(如b离子、y离子)和碎片离子质量误差容限。搜库软件比对实验谱图和理论谱图之后将给出带有打分值的肽段谱图匹配(peptide spectra match, PSM)其打分值标识出PSM的可信度。
如图中所示Best match是肽段VSTPNVSVVDLTCR,其打分值是5.6,相较于随后的打分值1.3和1.1是最高的。所以到此已经实现了这一条肽段的鉴定,将酶切后所有的肽段都鉴定之后根据酶切规律进行蛋白的组装,仍然用打分值来表征该组装蛋白可信度的高低,从而实现对蛋白的鉴定。
参考文献:
Alexey I. Nesvizhskii. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J Proteomics. 2010.

BioEngX
BioEngX---专属生物人的知识共享平台。您可以在这里分享实验经验、科研进展、行业资讯,以及招生招聘信息。点击阅读原文,加入我们!官方网站:http://www.bioengx.com/








