徐佳熹、孙媛媛、黄翰漾、刘鹭、黄昭宇、东楠、朱新彦、王楠、王佳慧、
杨希成、
李博康、李昶霖、蔡莹琛(港)、李伟(港)
、周逸(港)
、余克清(港)

资料来源:CDE,兴业证券经济与金融研究院整理
1. 百济神州
PD-1
第
6
项适应症上市申请获受理,治疗
NSCLC
3
月
6
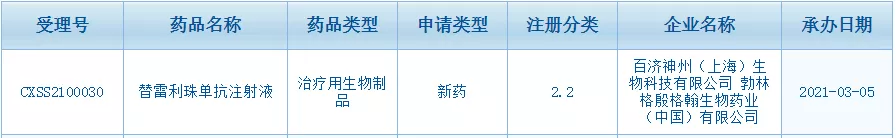
日,百济神州宣布,中国国家药品监督管理局(
NMPA
)药品审评中心(
CDE
)已受理其抗
PD-1
抗体百泽安
®
(替雷利珠单抗)用于治疗接受铂类化疗后出现疾病进展的二或三线局部晚期或转移性非小细胞肺癌(
NSCLC
)患者的新适应症上市申请。这是替雷利珠单抗的第
6
项上市申请。
资料来源:
CDE,兴业证券经济与金融研究院整理
此项
sBLA
是基于
RATIONALE 303
研究的中期分析结果。
RATIONALE 303
研究是一项随机、开放性、多中心的全球
III
期临床试验(
NCT03358875
),旨在评估百泽安
®
对比多西他赛用于治疗接受铂类化疗后出现疾病进展的二或三线局部晚期或转移性
NSCLC
患者的有效性和安全性。该试验的主要终点为在全部患者(意向治疗患者人群)中以及在
PD-L1
高表达患者中的
OS
;关键次要终点包括客观缓解率(
ORR
)、缓解持续时间(
DoR
)、无进展生存期(
PFS
)及安全性。该试验共在亚洲、欧洲、美洲和大洋洲的
10
个国家入组了
805
例患者,以
2:1
的比例随机至百泽安
®
试验臂或多西他赛试验臂。

资料来源:医药魔方,
CDE,兴业证券经济与金融研究院整理
百济神州于
2020
年
11
月宣布,经独立数据监查委员会(
DMC
)评估判断,
RATIONALE 303
临床试验在事先计划的中期分析中达到了
OS
这一主要终点。百泽安
®
的安全性数据与已知风险相符,未出现新的安全警示。百济神州预计在
2021
年上半年即将举行的一场医学会议上公布
RATIONALE 303
试验结果。
2. 安进
19
亿美元收购
FivePrime
3
月
4
日,安进宣布与
FivePrime Therapeutics
达成最终协议将以
38
美元
/
股的价格全现金收购后者股票对应股权总金额大约
19
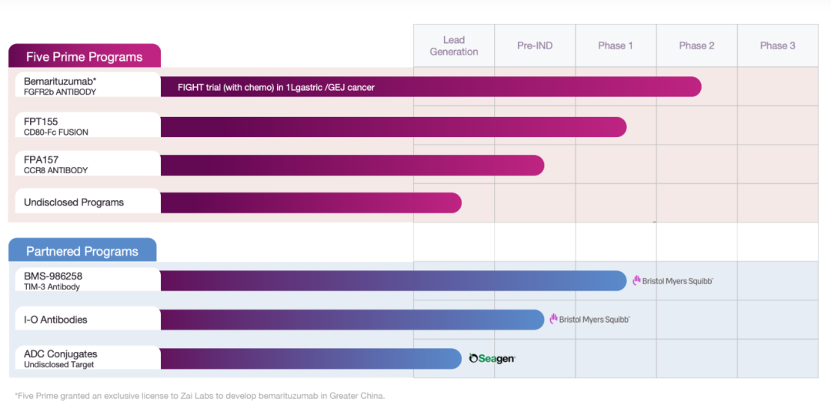
亿美元安进通过此次收购补强了自己的肿瘤创新药管线,包括获得了
first in class
的
FGFR2b
单抗
bemarituzumab
该药针对晚期胃癌或胃食管衔接部癌的临床开发已处于
III
期阶段。该交易预计在
2021
第二季度末完成。
FivePrime Therapeutics
管线
 资料来源:公司官网
,兴业证券经济与金融研究院整理
资料来源:公司官网
,兴业证券经济与金融研究院整理
在招募了
155
例患者的
II
期
FIGHT
研究中,
bemarituzumab
联合改良
FOLFOX6
方案一线治疗
FGFR2b+
、
HER2-
晚期胃癌及胃食管连接部腺癌患者,较安慰剂对照组显著改善了
PFS
(
9.5 vs 7.4
个月)、
OS
和
ORR
。进一步的分析还显示,肿瘤细胞的
FGFR2b
表达水平跟
bemarituzumab
的疗效有正向关系,支持了
FGFR2b
靶点的重要性以及
bemarituzumab
的抗癌活性。
成纤维细胞生长因子(
FGF
)
/
成纤维细胞生长因子受体(
FGFR
)通路与癌细胞的发育和生长有关。
FGFR2b
是
FGFR2
的剪接变体,发现与上皮起源的肿瘤中。
FIGHT
试验的数据表明,
HER2-
胃食管癌患者中大约
30%
存在
FGFR2b
过表达,在肺癌,乳腺癌,卵巢癌和其他癌症中也存在
FGFR2b
过表达。
3. 百奥泰终止
PD-1
单抗和
Trop2
抗体偶联药物项目临床开发
3
月
4
日,百奥泰公告宣布终止
BAT8003
(注射用重组人源化抗
Trop2
单克隆抗体
-
美登素偶联物)
和
BAT1306
(重组人源化抗
PD-1
单克隆抗体注射液)的临床开发。
BAT8003
是百奥泰自主开发的一种创新药物,是一种靶向
Trop2
的
ADC
药物,由糖基化修饰的重组人源化抗
Trop2
单克隆抗体通过定点偶联技术与
Batansine
进行共价连接而成,临床适应症是
Trop2
阳性晚期上皮癌。
BAT8003
于
2019
年
3
月开展
Trop2
阳性晚期上皮癌患者的
I
期临床试验,
在临床前的评估中,
BAT8003
已于临床前研究证明良好的耐受性及
PK
特征以及
良好疗效。考虑到当今
Trop 2 ADC
领域的市场格局变化,并且考虑到
BAT8003
与
BAT8001
在某些技术特征有类似(虽然
BAT8003
定点偶联,但是也用了
batansine
技术),存在较高的临床开发与市场风险。
BAT1306
是百奥泰开发的
PD-1
单抗,单药适应症为
EBV
相关性胃癌,目前已开展
BAT1306
联合
XELOX
一线治疗
EBV
相关
胃癌临床试验及
BAT8001
联合
BAT1306
二线治疗
HER2
阳性晚期实体瘤的临床
试验。百奥泰已终止
BAT8001
项目的推进,针对
BAT8001
联合
BAT1306
二线治疗
HER2
阳性晚期实体瘤的临床试验也因上述项目的终止而终止。从各家
PD-1
单抗的临床数据来看,预计未来
2-3
年,全球上市
PD-1
产品将可能超过
20
个,市场竞争日趋激烈。中国是
PD-1
竞争最激烈的地区,全球
154
个
PD-1
中有
85
个是由中国企业研发或合作开发,占比达到
55%
。
PD-1
全球和国内的
PD-1
单抗的研发赛道已经变得拥挤,繁多的研发竞品也加剧了对
CRO
公司的竞争,使得开发成本进一步加剧。
综合上述原因,鉴于
BAT8003
及
BAT1306
目前分别处于临床
I
期、
II
期,都属于较为早期阶段,后续开展临床实验还需要耗费公司大量人力财力。为合理
配置公司研发资源,聚焦研发管线中的优势项目,百奥泰审慎考量后决定终止上述项目的临床试验。
对于终止上述项目对于公司的损失,百奥泰坦诚除了
BAT8003
项目累计研发投入
6156.50
万元(截至
2020
年
12
月),
BAT1306
项目累计研发投入
5197.45
万元(截至
2020
年
12
月),也可能会导致公司在上述市场失去竞争地位。
百奥泰曾在
2
月
8
日公告终止了针对乳腺癌的
HER2 ADC
项目
BAT8001
的临床开发
,此前研发投入合计
2.26
亿元。加上此次终止的
2
个项目,累计研发投入损失为
3.4
亿元。
4. 礼来宣布旗下双重激动剂
tirzepatide
头对头
III
期研究成功
3
月
4
日,礼来宣布其开展的一项为期
40
周的
SURPASS-2
研究成功,该研究的顶线结果证明了全部
3
个剂量的
tirzepatide
在改善成人
2
型糖尿病患者的血糖水平和体重方面均优于诺和诺德旗下的明星药司美格鲁肽
1mg
。
SURPASS-2
试验是礼来大型
III
期
SURPASS
项目中迄今为止规模最大的一个,在全球范围内招募了
1879
例每日单独服用二甲双胍≥
1500mg
不能较好控制血糖的成人
2
型糖尿病患者(基线
HbA1c
水平
8.28
,基线体重
93.7kg
),按照
1:1:1:1
随机分组,在二甲双胍基础上给予每周
1
次
tirzepatide 5mg
,
10mg
,
15mg
或司美格鲁肽
1mg
,研究的主要终点是
tirzepatide 10mg
,
15mg
治疗组患者第
40
周的
HbA1c
水平较基线的改善程度不劣于司美格鲁肽。关键次要终点包括:
1
)
tirzepatide 5mg
组的降糖效果非劣效;
2
)
HbA1c
水平和体重较基线改善更明显以及
tirzepatide 3
个剂量组
HbA1c
<
7
的患者比例更高;
3
)
tirzepatide 10mg
,
15mg
治疗组患者
HbA1c
<
5.7
的患者比例更高。
按疗效估量的结果显示,
3
个剂量
tirzepatide
治疗组在降低血糖和减轻体重方面的效果均好于司美格鲁肽,并且
HbA1c
<
7
的患者比例均高于司美格鲁肽(
10mg
和
15mg
剂量组具有统计学显著性差异,
5mg
组同样具有统计学显著性差异)。
资料来源:公司官网
,FDA,兴业证券经济与金融研究院整理
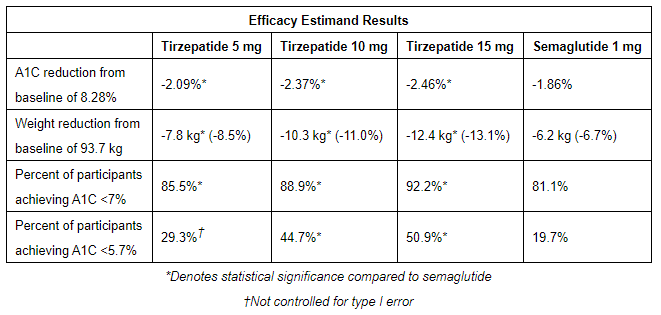
如果从治疗方案估量的具体数据来看,
Tirzepatide 5mg
,
10mg
,
15mg
或司美格鲁肽
1mg
组
HbA1c
水平较基线分别降低
2.01%
,
2.24%
,
2.30%
,
1.86%
;体重分别降低
7.6kg
,
9.3kg
,
11.2kg
,
5.7kg
;
HbA1c
<
7
的患者比例分别为
82.0%
,
85.6%
,
86.2%
,
79.0%
;
HbA1c
<
5.7
的患者比例分别为
27.1%
,
39.8%
,
45.7%
,
18.9%
;
安全性方面,
SURPASS-2
试验报道的数据与之前研究以及
GLP-1R
激动剂的情况一致。
Tirzepatide 5mg
,
10mg
,
15mg
或司美格鲁肽
1mg
最常见的不良事件均为胃肠道相关不良事件,包括恶心(
17.4%
,
19.2%
,
22.1%
,
17.9%
)、腹泻(
13.2%
,
16.4%
,
13.8%
,
11.5%
)、呕吐(
5.7%
,
8.5%
,
9.8%
,
8.3%
),因不良事件终止治疗的患者比例分别为
5.1%
,
7.7%
,
7.9%
和
3.8%
。低血糖(<
54mg/dL
)的发生率分别为
0.6%
,
0.2%
,
1.7%
和
0.4%
。
Tirzepatide
是一种新型的每周
1
次的葡萄糖依赖性促胰岛素多肽(
GIP
)和胰高血糖素样肽
-1
(
GLP-1
)双重激动剂,可将两种肠促胰岛素的作用整合到了一个分子中。临床前研究表明,
GIP
可通过降低食物摄入和增加耗能来减轻体重,与
GLP-1
受体激动剂结合,可能对患者血糖和体重产生更大影响。
5. 首个
Claudin18.2/PD-L1
双抗获批临床
3
月
4
日,国家药监局药品审评中心
CDE
官网公示启愈生物的
Q-1802
冻干粉针新药临床试验申请获得临床默示许可适应症为晚期实体瘤。
Q-1802
是中国首个申报临床的
Claudin18.2/PD-L1
双特异性抗体。
Q-1802
是启愈生物利用其抗体工程技术平台自主开发并具有自主知识产权的可以同时靶向
PD-L1
及
Claudin18.2
的双特异性抗体。
Q-1802
,体内动物药效验证了上述机制,并显示
Q-1802
药效优于
PD-L1
抗体和
Claudin18.2
抗体的联用。
Q-1802
可利用
Claudin18.2
抗体将
PD-L1
抗体特异性地靶向到肿瘤组织,显著降低系统暴露量,降低副作用。
就在
3
月
1
日,美国
FDA
也已批准
Q-1802
的
IND
申请,是全球首个通过美国
FDA IND
的
Claudin18.2/PD-L1
双特异性抗体。
6. 贝达药业三代
EGFR
抑制剂申报上市,四代获批临床
3
月
4
日,
CDE
官网显示,贝达药业
1
类新药甲磺酸贝福替尼胶囊(
BPI-D0316
)上市申请正式获
CDE
受理。拟用于治疗既往使用
EGFR-TKI
耐药后产生
T790M
突变的局部晚期或转移性非小细胞肺癌。
甲磺酸贝福替尼胶囊是一种第三代
EGFR-TKI
,含有新的结构明确的、具有药理作用的化合物,属于“境内外均未上市的创新药”,其注册分类为化学药品
1
类,其有望用于治疗具有
T790M
突变及其他突变的
EGFR
阳性肺癌患者。
研究结果显示贝福替尼胶囊
75-100mg
治疗既往使用
EGFR-TKI
耐药后产生
T790M
突变的局部晚期或转移性
NSCLC
患者,经独立评审委员会(
IRC
)评估的客观缓解率(
ORR
)为
64.8%
,疾病控制率(
DCR
)
为
95.2%
。颅内客观缓解率(
iORR
)为
52.9%
,颅内疾病控制率(
iDCR
)为
97.1%
,
提示
贝福替尼胶囊对颅内病灶同样具有较好的疗效。从数值上看,贝福替尼胶囊的整体疗效和颅内疗效均与其他三代
EGFR-TKI
基本一致。无进展生存期
(
PFS
)数据尚未成熟。
安全性方面,贝福替尼胶囊安全耐受性良好,尽管大部分患者均会发生不良反应,但大多为
1
级或
2
级,最常见的不良反应为血小板减少症、头痛、白细胞计数降低等。最常见的
3
级及以上不良反应为血小板减少症,不良反应经暂停用药和
/
或对症治疗等可恢复或缓解,提示
BPI-D0316
胶囊的不良反应可耐受可控、转归良好。
此外,
3
月
3
日公司旗下的第四代
EGFR-TKI BPI-361175
也已获批临床。
7. 葛兰素史克「多替拉韦拉米夫定片」在华获批上市
3
月
3
日,国家药监局官网显示,葛兰素史克多替拉韦拉米夫定片已正式获
NMPA
批准上市,用于治疗用于治疗感染人类免疫缺陷病毒
1
型
(
HIV-1
)
的成人和
12
岁以上青少年(体重至少
40
公斤),且对整合酶抑制剂或拉米夫定无已知或可疑耐药患者。
多替拉韦拉米夫定片(
Dovato
)由
GSK
旗下的
ViiV Healthcare
研发,于
2019/4/8
获得
FDA
批准上市,
2019/7/3
获得欧盟批准,
2020
年销售额为
4.83
亿美元。这是一种抗
HIV
感染的双药疗法,有效性不劣于指南推荐的三药疗法,同时降低终生累积药物暴露量和潜在长期毒性。也是
FDA
批准的首个用于既往从未接受过治疗的成人
HIV-1
感染的双药固定剂量完整疗法。
Dovato
由固定剂量的多替拉韦和拉米夫定组成,其中,多替拉韦是一种
HIV
整合酶抑制剂,能够通过阻止病毒
DNA
整合至人体免疫细胞的遗传物质来阻断
HIV
的复制。拉米夫定是一种核苷类逆转录酶抑制剂,常与其他抗逆转录病毒药物联合使用,用于
HIV
感染的治疗。
一项代号为
ANGO
的开放标签、多中心、
III
期研究评估了处于病毒学抑制阶段(
HIV-1 RNA <50
拷贝
/mL
)的
HIV-1
感染成人患者换药为
Dovato
的疗效、安全性和耐受性。研究结果显示,第
48
周时,接受
Dovato
治疗患者
HIV-1 RNA
≥
50
拷贝
/mL
患者比例与继续接受替诺福韦艾拉酚胺(
TAF
)为基础
3
联或
4
联治疗方案患者比例无显著性差异(
0.3%
(
1/369
)
vs 0.5%
(
2/372
))
,
达到非劣效性标准,治疗过程中无病毒学失败或紧急耐药性报告。
8. 礼来
JAK
抑制剂
III
期研究成功,用于脱发治疗
3
月
3
日,礼来和
Incyte
联合宣布
JAK
抑制剂巴瑞替尼治疗成人重度斑秃(
AA
)的
III
期研究(
BRAVE-AA2
)达到积极顶线结果。两个剂量组巴瑞替尼
(
2mg
和
4mg
,每日
1
次)在第
36
周时均达到主要疗效终点,与安慰剂相比,头皮毛发再生有了统计学上的显著改善。该研究的详细结果将在即将召开的医学会议上公布。
巴瑞替尼是首个在治疗
AA
的
III
期试验中证明可促使毛发再生的
JAK
抑制剂,目前还没有被
FDA
批准治疗斑秃的药物
。
巴瑞替尼在
BRAVE-AA2
研究中的安全性结果与在类风湿关节炎(
RA
)和特应性皮炎(
AD
)患者中的既定安全性一致。无死亡、主要不良心血管事件(
MACE
)或静脉血栓栓塞事件(
VTEs
)的报告。
BRAVE-AA2
研究是一项多中心、随机、双盲、安慰剂对照研究,共纳入了
546
例头皮脱发≥
50%
(
SALT
评分≥
50
)和严重
AA
症持续至少
6
个月但不超过
8
年患者。包含了来自中国、韩国、日本、巴西、澳大利亚、阿根廷、美国等世界上多个国家的患者人群。
RAVE-AA2
研究是首个在
AA
患者中获得阳性结果的
III
期研究,其用于治疗
AA
中另一项
III
期研究数据也将于今年上半年获得。
巴瑞替尼是一种口服
JAK
抑制剂,由
Incyte
开发并授权给礼来,商品名为
Olumiant
,目前已在包括美国在内的全球
70
多个国家获批用于治疗成人中重度活动性类风湿性关节炎(
RA
)。在欧盟和日本获批用于治疗成人中重度特应性皮炎(
AD
)。此外,巴西替尼正在开展治疗系统性红斑狼疮(
SLE
)、幼年特发性关节炎(
JIA
)和
COVID-19
的研究。
巴瑞替尼片已于
2019
年
6
月在中国获批上市用于对一种或多种改善病情抗风湿药(
DMARD
)疗效不佳或不耐受的中重度活动性类风湿关节炎成年患者。礼来也已在中国开展巴瑞替尼片治疗重度及极重度成人斑秃(
AA
)的
III
期研究。
9. 君实生物向
FDA
滚动提交
PD1
单抗上市申请
3
月
3
日君实生物宣布向美国
FDA
滚动提交了特瑞普利单抗用于治疗复发或转移性鼻咽癌的生物制品许可申请
BLA2020
年
9
月特瑞普利单抗用于治疗复发或转移性鼻咽癌获得
FDA
突破性疗法认定由于获得该突破性疗法认定特瑞普利单抗治疗鼻咽癌的
BLA
可向
FDA
滚动提交并获得滚动审评
Rolling Review
。
滚动审评是指药企在申请新药上市许可时可以将申报文件分批次提交
FDA
进行审评而无需等待申报文
件全部完成后才向
FDA
提交申请此举可缩短新药的审评周期特瑞普利单抗成为首个向
FDA
提交
BLA
的国产抗
PD-1
单抗。
特瑞普利单抗是中国首个批准上市的以
PD-1
为靶点的国产单抗药物且至今已在中美等多国开展了覆盖超过
15
个适应症的
30
多项临床研究
2018
年
12
月
17
日特瑞普利单抗获得国家药品监督管理局有条件批准上市用于既往接受全身系统治疗失败的不可切除或转移性黑色素瘤的治疗并获得
2019
年和
2020
年中国临床肿瘤学会(
CSCO
)黑色素瘤诊疗指南推荐
2021
年
2
月特瑞普利单抗治疗既往接受过二线及以上系统治疗失败的复发
/
转移性鼻咽癌患者的新适应症上市申请获得国家药监局有条件批准。
特瑞普利单抗用于二线治疗转移性尿路上皮癌的新适应症上市申请已于
2020
年
5
月获得国家药监局受理并于
2020
年
7
月被国家药监局纳入优先审评程序
2020
年
12
月特瑞普利单抗注射液成功通过国家医保谈判被纳入新版国家医保目录
2021
年
2
月特瑞普利单抗联合化疗用于晚期一线未接受过系统性治疗的复发转移性鼻咽癌的新适应症上市申请获得国家药监局受理目前特瑞普利单抗已在黏膜黑色素瘤鼻咽癌软组织肉瘤领域获得
FDA
授予
1
项突破性疗法认定
1
项快速通道认定和
3
项孤儿药资格认定。
10.
思路迪
国内首个
AXL
抑制剂申报临床
3
月
3
日,思路迪
AXL
抑制剂
3D-229
注射液临床试验申请正式获
CDE
受理
,
是国内首个申报临床的
AXL
抑制剂。
3D-229
(
AVB-500
),由美国
Aravive
公司开发,
2020
年
11
月
11
日,思路迪以总计
2.19
亿美元从
Aravive
公司获得了
3D-229
大中华区肿瘤领域的独家开发和商业化权利。
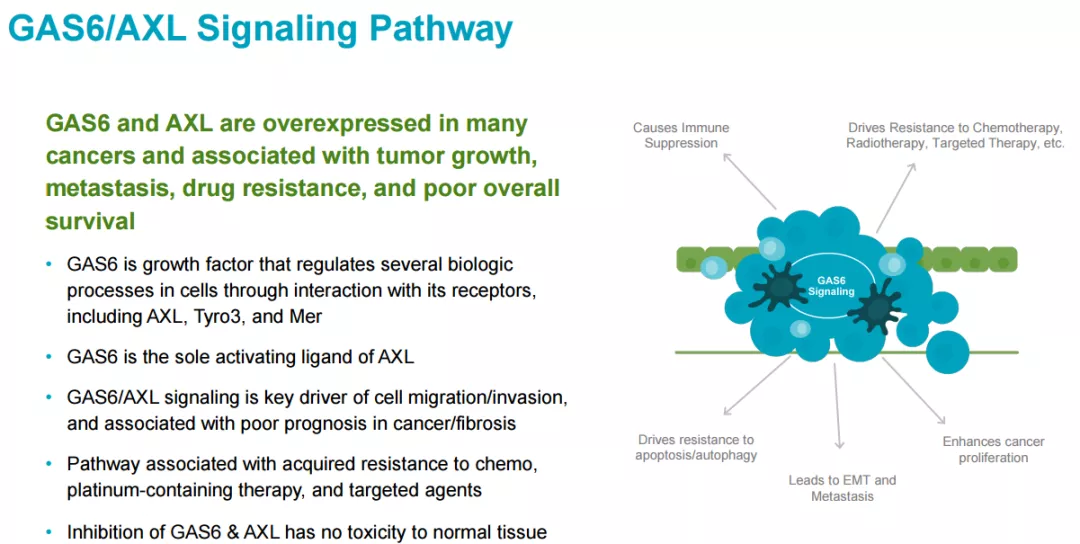
GAS6-AXL
信号通路是促进肿瘤生长及转移、肿瘤免疫逃逸与药物耐受的关键通路。
AXL
及其配体
GAS6
在许多恶性肿瘤中都有高表达和活化,如膀胱癌(
>45%
),肾癌(
>70%
),胰腺癌(
70%
),乳腺癌(
>75%
),肺癌(
18-48%
),卵巢癌(
>70%
)以及前列腺癌等。
GAS6
是
AXL
唯一的激活配体。
3D-229
(
AVB-500
)是一款诱导融合蛋白,可对
GAS6/TAM
通路中的配体
GAS6
进行有选择性的高亲和力中和。
资料来源:公司官网
,兴业证券经济与金融研究院整理
AVB-500
的优点是对
GAS6
配体据有高度亲合力,从而解决了靶向
AXL
的
TKI
抑制剂低选择性,导致的脱靶毒性,肿瘤细胞耐药性问题和单抗隆抗体低亲和力,不足以破坏天然
GAS6/TAM
之间相互作用问题。

资料来源:公司官网
,兴业证券经济与金融研究院整理
此前
AVB-500
在一项针对铂耐药性卵巢癌(
PROC
)患者的
Ib
期临床试验显示出了潜在疗效。
10 mg/kg
剂量组
37
例疗效可评估患者整体
ORR
为
21.6%
(
8/37
)。
15mg/kg
剂量组
5
例疗效可评估患者中,
1
例达到
CR
,
2
例部分缓解(
PR
),
2
例疾病稳定(
SD
)。
20mg/kg
剂量组
7
例疗效可评估患者中,
1
例达到
PR
,
1
例
SD, 5
例疾病进展(
PD
)。
目前
AVB-500
针对肾细胞癌、尿路上皮癌的临床试验也正在进行中,并已在铂耐药复发性卵巢癌适应症上获得
FDA
授予的快速通道资格,该适应症的关键临床试验也即将展开。

资料来源:公司官网
,FDA,兴业证券经济与金融研究院整理
11. SMO
抑制剂上市申请获
CDE
优先审评,治疗基底细胞癌
今日,上海济煜医药
/Sun Pharma
磷酸索尼德吉胶囊(商品名:
Odomzo
)上市申请被
CDE
纳入优先审评并开始公示,用于手术或放疗后复发的局部晚期基底细胞癌(
BCC
)成年患者,或不宜手术或放疗的患者。
磷酸索尼德吉是诺华开发的一款口服性
Smoothened
(
SMO
)抑制剂。
2017
年印度太阳制药与诺华签署了一份许可协议,获得了
Odomzo
全球范围商业化权利。
SMO
是一种跨膜蛋白,是
Hedgehog
(
Hh
)信号传导通路的一部分,该通路在干细胞维持、组织修复、晚期基底细胞癌中发挥关键作用。
Odomzo
通过调控
Hedgehog
信号通路,从而阻止或减少癌变的发展。
磷酸索尼德吉最早于
2015
年
7
月
24
日获得美国食品药品管理局(
FDA
)
批准上市
,用于治疗手术或放疗后复发的局部晚期基底细胞癌,或不能手术或放疗的基底细胞癌患者。
2015
年
8
月
20
日在欧盟获准上市。
Odomzo
获
FDA
批准是基于一项针对局部晚期基底细胞癌(
laBCC
)
(
n=194
)或转移性基底细胞癌(
mBCC
)
(
n=36
)患者多中心、双盲、多队列临床研究((
BOLT, NCT01327053
)。患者按
2:1
比例随机接受口服
ODOMZO 800 mg
或
200 mg
,每日
1
次治疗。研究结果显示,第
30
个月时,随机接受每日
200mg ODOMZO
治疗
laBCC
患者
ORR
为
56%
,
CR
率为
21%
。中位缓解持续时间为
26.1
个月。
基底细胞癌(
BCC
)
是常见皮肤癌,占所有非黑色素瘤病例的
80%
,该病晚期能严重损毁外形并危及生命。
BCC
发病率以每年约
10%
的速率上升。晚期
BCC
目前的治疗选择非常有限。
12. 艾力斯三代
EGFR-TKI
甲磺酸伏美替尼获
NMPA
批准上市
3
月
3
日,国家局官网显示,艾力斯医药第三代表皮生长因子受体酪氨酸激酶抑制剂(
EGFR-TKI
)甲磺酸伏美替尼已获
NMPA
批准上市,用于治疗
EGFR T790M
突变阳性的局部晚期或转移性非小细胞肺癌(
NSCLC
)。
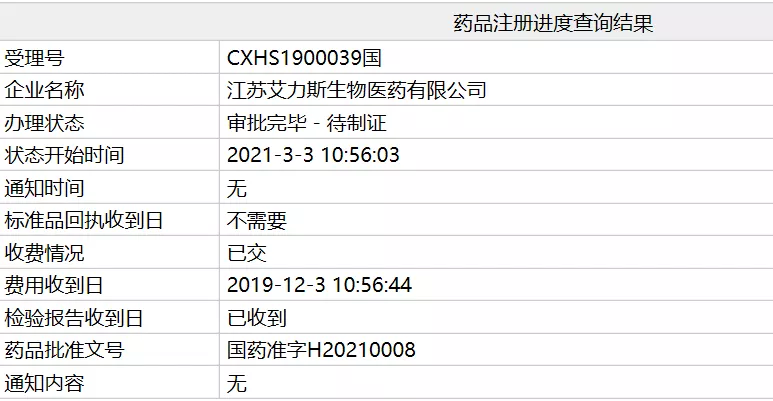
资料来源: CDE,医药魔方,
兴业证券经济与金融研究院整理
我国
NSCLC
患者
EGFR
敏感突变率约为
50%
,这类患者常规使用第一
/
二代
EGFR-TKI
治疗。但多数患者在接受治疗一年左右出现耐药和疾病进展,其中约
60%
的患者为
EGFR T790M
耐药突变,这类患者目前最有效的治疗药物为第三代
EGFR-TKI
。
一项代号为
ALSC003
的
IIb
期多中心单臂研究评估了伏美替尼治疗
T790M
突变
NSCLC
患者的疗效和安全性。研究纳入局部晚期或转移性经一
/
二代
EGFR TKI
治疗后进展或初治
T790M
突变
NSCLC
患者,接受伏美替尼(
80mg
,一日两次),主要研究终点为
ORR
,次要终点包括
DCR
、
PFS
和
OS
。
2018
年
6
月至
2018
年
12
月期间,纳入
220
例患者,
212
例患者为
IV
期,截止
2019
年
4
月
12
日,患者
ORR
为
73.6%
,预估
6
周和
12
周
DCR
率为
87.3%
和
82.3%
,中位
PFS
为
7.6
个月,中位
OS
和
DoR
均未达到。
艾力斯医药于
2019
年
11
月向
NMPA
提交了伏美替尼附条件批准上市申请,并以“具有明显治疗优势创新药”为由被
CDE
纳入优先审评。










