
原文链接:
https://www.nature.com/articles/s41586-019-1472-0
CCCTC-结合因子(CCCTC-binding factor,CTCF)能够锚定DNA环,使基因组分布于不同的结构域,通过促进或限制基因与调控元件之间的相互作用进而控制基因表达
1
。在肿瘤细胞中,体细胞突变
2
或DNA超甲基化
3
破坏CTCF在特定位点的结合,引起DNA环锚定缺失,激活原癌基因表达。相反,
CTCF
生殖细胞特异性的旁系同源物
BORIS
(brother of the regulator of imprinted sites,BORIS)在不同肿瘤中都是过表达的
4
,但是其在恶性表型中的作用尚不清楚。在本文中,研究工作者发现
BORIS
异常上调促进
ALK
突变、
MYCN
扩增型神经母细胞瘤细胞中染色质相互作用从而使这些细胞具有
ALK
抑制的抗性
。在获得抗性的过程中,这些细胞被重编程为一种不同的细胞状态,最初
MYCN
表达缺失,随后
BORIS
过表达。由此产生的BROIS调控染色质环的改变导致超增强子的形成,驱动转录因子的异位表达,最终决定了耐药表型。因此,本研究揭示BORIS能促进调控染色质相互作用,维持特定癌症表型。
1.神经母细胞瘤靶向治疗耐药与转录重编程以及对MYCN向BORIS依赖的转变有关
CTCF在健康组织和肿瘤细胞中均有表达,而BORIS通常表达于睾丸
5
和胚胎干细胞
6
。然而,BORIS在肿瘤中异常表达常与高位风险相关(例如治疗抗性)
4
。研究工作者在先前研究中发现扩增的
MYCN
7
和突变ALK(F1174L)
8
促进神经母细胞瘤中BORIS高表达,抵抗ALK抑制。如图1a,Kelly人神经母细胞瘤细胞暴露于浓度增加的ALK抑制剂TAE684直至其获得稳定的细胞抗性。获得稳定的耐药性不仅与ALK磷酸化的丧失相一致(这表明细胞不再需要激活这种受体酪氨酸激酶来维持其致癌特性),而且还与缺乏其他常见的耐药性激发因子相一致(补充材料图2a,e-h)。然而,如图1b,比较TAE684敏感性和抗性细胞中基因表达谱可知,抗性细胞中大多数基因表达下调,但
与神经母细胞瘤细胞无关的一个转录因子子集显著上调
9
。因此,研究工作者认为
耐药细胞可能在耐药过程中经历了转录重编程
。为确定抗性发展的动态过程,研究工作者对敏感状态、过渡状态和完全抗性状态的细胞进行了单细胞测序(single-cell RNA sequencing,scRNA-seq)分析。如图1c,主成分分析(principal component analysis,PCA)提示
细胞从敏感状态向完全耐药状态的逐步过渡
。如图1d,研究工作者通过细胞抗性获得过程中差异表达转录因子的拟时间分析发现,
最初
MYCN
表达缺失且持续低表达于稳定的耐药细胞
。为理解这个意想不到的结果,研究工作者分析了细胞中
MYCN
的状态,发现
MYCN
基因组扩增仍然存在,但是MYCN基因位点发生了表观沉默抑制(补充材料图3f-3g)。这种状态与基因组范围内MYCN在DNA上的结合降低是一致的(图1e)。与转录活性缺失一致,抗性细胞不再依赖于MYCN存活,而敏感性细胞在敲减MYCN后发生凋亡(补充材料图图3i)。此外,如图1d,
在细胞从敏感型向抗性转变的过程中,随着
BORIS
表达升高,神经发育的标志物
SOX2
和
SOX9
表达逐渐增加
。有趣的是,BORIS过表达与其启动子低甲基化相一致(补充材料4a-4b),这种情况同样存在于对TAE684或CDK12抑制剂E9产生抗性的其他神经母细胞瘤细胞系,提示该发现并非局限于单一细胞系或抑制剂。
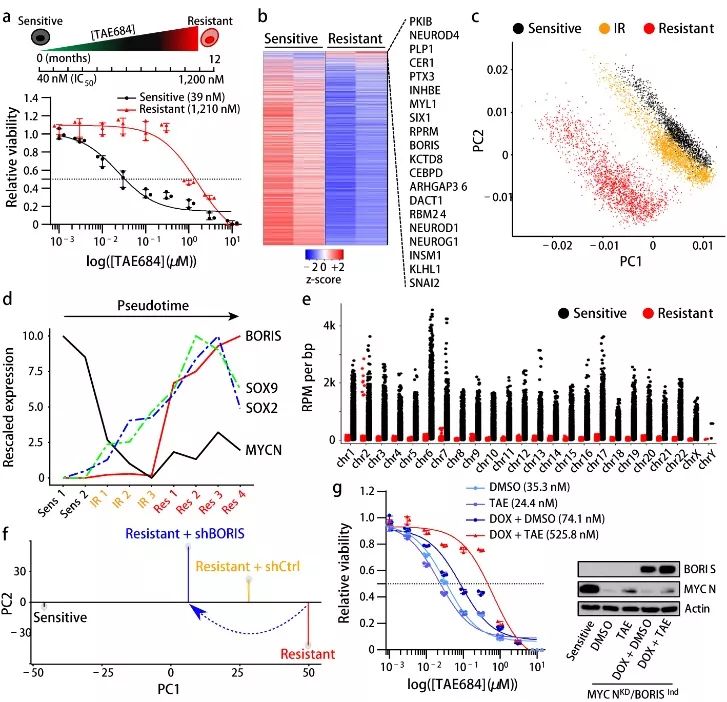
图1 神经母细胞瘤耐药性产生与对MYCN和BORIS表达改变的依赖相关
为明确BORIS在细胞抗性表型中的作用,研究工作者在抗性细胞中敲减BORIS,如图1f,抗性细胞部分逆转成敏感性细胞,同时伴随MYCN和ALK表达增加(补充材料图5a-5c)。然而,这一结果并不能够维持细胞生长,因为在抗性细胞中敲减BORIS降低细胞的生存能力(补充材料图5d-5e),提示从MYCN到BORIS依赖性的转变具有稳定的阻力。这种转变与细胞生长动力学的变化有关:从高度增殖、MYCN过表达的敏感状态到完全耐药细胞中部分逆转的中间、慢循环表型,与BORIS表达相一致(补充数据图5f-5h)。如图1g,
在
ALK
抑制的情况下,敏感细胞产生耐药性需要
MYCN
表达下调和
BORIS
过表达
。因此,神经母细胞瘤细胞对ALK抑制的耐药性是一个多步骤的过程,促进其对扩增MYCN(主要致癌刺激)的依赖向BORIS过表达的转变,提示
BORIS
在细胞耐药性中起着重要的作用
。
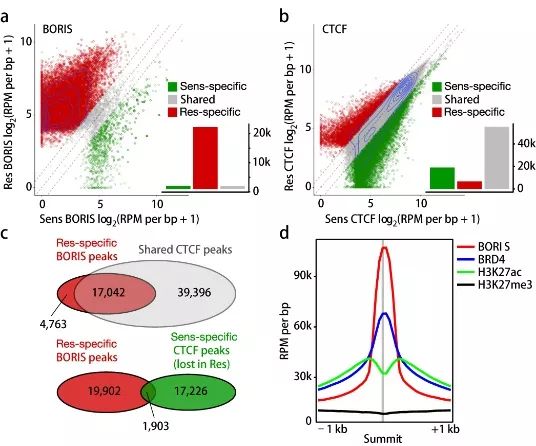
图2 BORIS过表达与其在耐药细胞中染色质占用的增加有关
2. BORIS过表达与其在耐药细胞中染色质占用的增加有关
为进一步确定异常表达的BORIS是否影响其在染色质上的占有率,研究工作者对敏感性细胞和抗性细胞进行了ChIP-seq,如图2a,BORIS在敏感性细胞和抗性细胞中染色质的占有峰分别为2211和22891,即
BORIS
在抗性细胞中染色质占有增加
。相反,CTCF在敏感性细胞和抗性细胞中染色质的占有峰分别为75567和63246,即
CTCF
在抗性细胞中染色质占有降低
。有趣的是,如图2c,相当大一部分(n = 17042;78%)耐药细胞特有的BORIS峰与两种细胞共享的CTCF峰重叠。然而,只有一小部分((n = 1,903;8.7%)与CTCF峰重叠的BORIS是抗性细胞所特有的,提示
BORIS
并不能替代抗性细胞中的
CTCF
。此外,在抗性细胞中,BORIS倾向于结合在基因调控元件增强子和启动子(补充材料图6d-6e),与它与开放染色质区域(H3K27ac)结合的倾向相一致(图2d)。因此,
BORIS
过表达与其在耐药细胞中染色质占用的增加有关
。
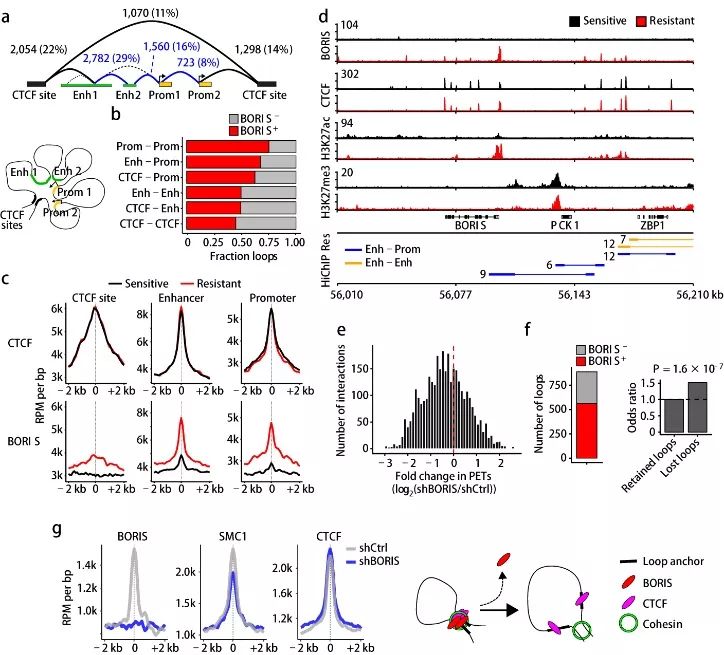
图3
BORIS
促进耐
药细胞中
新的染色质相互作用
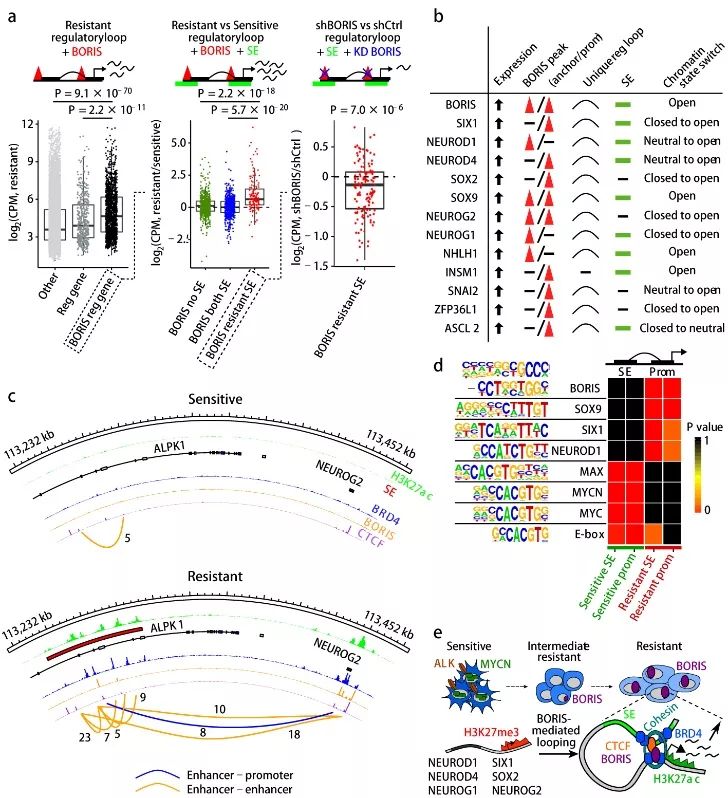
图4 BORIS调控的染色质重塑是维持细胞耐药状态的表型开关
3. BORIS促进耐药细胞中新的染色质相互作用
在耐药细胞中,BORIS结合的基因组区域与活跃的染色质特征相关,这提示
BORIS
可能像
CTCF
和内聚蛋白一样,通过染色质环调控基因表达
。因此,研究工作者在敏感性和抗性细胞中首先使用以内聚蛋白(SMC1A)为基础的高通量染色体构象捕获技术,然后再进行染色质免疫沉淀。如图3a,基于相关环锚定在基因组上位置,研究工作者确定了六种不同的相互作用:三种较长的与CTCF位点相互作用的染色质环和三种较短的与调控元件相互作用的染色质环。如图3b,BORIS结合与染色质环锚点重叠分析揭示抗性细胞中大多数(56%)相互作用是BORIS阳性的。值得注意的是,
BORIS
在锚点上的富集改变与调控区域相关,而
CTCF
并未改变
(图3c-3d)。事实上,BORIS在未结合CTCF的锚点单独结合就足以在耐药细胞中产生新的相互作用(补充材料图7d)。为确定抗性细胞中新形成的相互作用是否由BORIS介导,研究工作者分析了敲减BORIS后的染色质环构象。敲减BORIS使耐药细胞中特有的调控相互作用呈现出全球性的损失(图3e),超过四分之一的相互作用消失了,其中63%是BORIS阳性的描点(图3f)。此外,敲减BORIS降低SMC1A的结合,并未显著影响CTCF的结合(图3g)。因此,
BORIS
是抗性细胞中形成染色质环形成的关键因子
。
4. BORIS调控的染色质重塑是维持细胞耐药状态的表型开关
如图4a,与BORIS阴性染色质环的基因或不存在新的相互作用的基因相比,
新的
BORIS
阳性染色质环的基因表达水平显著升高
。在健康细胞和肿瘤细胞中确定细胞身份的基因经常受超级增强子调控
10
,研究工作者分析了细胞中超级增强子的分布,发现抗性细胞中特有的超级增强子富集于BORIS阳性的染色质环(补充材料图8a-8c)。这些超级增强子的存在与它们的相关基因在耐药细胞和敏感细胞中的高表达显著相关(图4a)。这些抗性细胞中BORIS阳性的超级增强子基因主要是调控紧密或中性状态染色质向开放构象转变的基因(补充材料图8d-8e)。敲减BORIS降低BORIS阳性染色质环基因的表达,特别是与抗性相关的超级增强子基因(图4a和补充材料图8f)。这些结果提示
BORIS
介导的染色质环改变导致新形成的超级增强子与其靶基因相互作用,从而促进其表达
。
接着,研究工作者通过分析融合基因表达、BORIS介导的染色质环、超级增强子分布和染色质状态等最终确定了89个与细胞抗性相关的受BORIS调控的基因(补充材料表),包括13个转录因子,其在早期神经发育中高表达且与决定细胞命运
11
(图4b-4c和补充材料8g)。这些促神经的转录因子表达与BORIS在耐药细胞中的表达一致,依赖于BORIS介导的染色质环,因为敲减BORIS导致了它们的下调(补充材料图8h-8i)。此外,研究工作者对这些转录因子的结合位点进行分析,发现在抗性细胞中几个转录因子和BORIS富集于高表达基因的调控区域,而在敏感性细胞中则主要受MYC、MYCN 、MAX E-box 和E-box-like motifs调控(图4d)。因此,
BORIS
诱导染色质重塑建立新的转录因子调控网络,维持细胞抗性状态
。
在本文中,研究工作者利用对ALK抑制剂敏感性和抗性的神经母细胞瘤细胞揭示了CTCF旁系同源物BORIS通过促进DNA相互作用产生耐药性的分子机制,如图4e,MYCN和ALK表达在敏感性细胞中,而受BORIS调控的特定转录因子(SIX1和SOX2等)则因紧密的染色质结构(H3K27me3)处于表达抑制状态;在敏感性状态向抗性状态过渡中,BORIS表达增加,介导染色质环形成并与超级增强子相互作用,使染色质处于开放状态(H3K27ac),促进受BORIS调控转录因子表达,建立受特定转录因子调控的基因表达网络,维持细胞的耐药性。
1. Dixon, J.R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions.
Nature
485
, 376-80 (2012).
2. Hnisz, D. et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods.
Science
351
, 1454-1458 (2016).
3. Flavahan, W.A. et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas.
Nature
529
, 110-4 (2016).
4. Martin-Kleiner, I. BORIS in human cancers -- a review.
Eur J Cancer
48
, 929-35 (2012).
5. Loukinov, D.I. et al. BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events, shares the same 11-zinc-finger domain with CTCF, the insulator protein involved in reading imprinting marks in the soma.
Proc Natl Acad Sci U S A
99
, 6806-11 (2002).
6. Monk, M., Hitchins, M. & Hawes, S. Differential expression of the embryo/cancer gene ECSA(DPPA2), the cancer/testis gene BORIS and the pluripotency structural gene OCT4, in human preimplantation development.
Mol Hum Reprod
14
, 347-55 (2008).
7. Brodeur, G.M., Seeger, R.C., Schwab, M., Varmus, H.E. & Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage.
Science
224
, 1121-4 (1984).
8. George, R.E. et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma.
Nature
455
, 975-8 (2008).
9. Durbin, A.D. et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry.
Nat Genet
50
, 1240-1246 (2018).
10. Whyte, W.A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes.
Cell
153
, 307-19 (2013).
11. Kumar, J.P. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease.
Cell Mol Life Sci
66
, 565-83 (2009).










