系列教程往期回顾:
Gromacs教程1-水
原文:James (Wes) Barnett
在本教程中,我将向您展示如何在一个盒子的TIP4PEW水中创建一个包含一个OPLS力场甲烷的系统。
设置
和以前一样,我们需要一个结构文件,一个拓扑文件和参数文件。我们将使用GROMACS工具
gmx pdb2gmx
从pdb文件生成拓扑。
用pdb2gmx设置残基
对于这个分子,我们将使用OPLS力场。 力场位于顶层力场目录(可能是
/usr/
share
/
gromacs
/
top
或类似的东西)。
如果您不确定您的GROMACS安装位置在哪里,可以使用如下命令:
$ echo $ GMXPREFIX
如果您sourcing 了GROMACS配置文件,则会为您提供安装位置。在该目录中找到
share
/
gromacs
/
top
并进入它(例如,如果GMXPREFIX是
/
usr
,则转到
/usr/
share
/
gromacs
/
top
)。或者你可以简单地转到
$GMXDATA
/
top
。
我们来看看force field目录的内容:
$ cd oplsaa.ff
$ ls
你会看到几个文件,但我们现在只对其中的一些文件感兴趣。注意
forcefield
.
itp
。这是模拟中使用的主要文件。在里面你会看到一个[
defaults
]部分以及包含两个其他文件 - 一个用于键联系,另一个用于非键联系。我们也对
atomtypes
.
atp
感兴趣,它给出了难以理解的
opls _
####
术语的描述以及
aminoacids
.
rtp
,它们给出了用于
gmx pdb2gmx
命令能够识别的氨基酸残基列表。
用你的文本编辑器打开
atomtypes
.
atp
,比如用vim打开它:
$ vim atomtypes.atp
转到
opls_138
的行。请注意,注释是否为
alkane CH4
。但是,请注意第二列中的mass - 这只是CH4组的碳,所以我们也需要氢。这是一个“全原子”模型 - 每个原子都被表示出来。相应的氢是
opls_140
。您可能还需要查看OPLS force field 论文的支持信息。论文中的参数应该与我们刚刚看到的参数相匹配。现在记下这两种原子类型并关闭文件。
让我们来看看这两种原子类型的ffnonbonded.itp:
$ grep opls_138 ffnonbonded.itp
$ grep opls_140 ffnonbonded.itp
在这里,我们看到原子类型的名称,键类型,质量,电荷,ptype,sigma和epsilon。记下每个类型的电荷 - 我们将需要它来构建我们新的氨基酸残基。作为一个方面说明,
ffbonded
.
itp
将使用键类型,键类型和二面角类型。
在继续之前,您可能希望将您的顶级力场目录复制到别的地方,例如您的主目录,因为我们将修改它并添加一些文件。将其复制到您的主目录中:
$ cp -r $GMXDATA/top $HOME/GMXLIB
你可能需要root权限执行它。现在将
$ GMXLIB
环境变量更改为:
$ export GMXLIB = $ HOME/GMXLIB
将上述内容添加到
.
bash_profile
中以使其成为永久性的,然后进行执行:
$ cd $ GMXLIB
您现在处于刚刚拷贝的副本中,所有模拟将使用该目录而不是GROMACS默认目录中提供的目录。
现在进入
oplsaa
.
ff
并打开
aminoacids
.
rtp
。您会注意到文件中已有多个残基。我们将为我们的甲烷添加一个名为
methane
.
rtp
的新文件,其中包含我们称之为CH4的残基。关闭
aminoacids
.
rtp
。我们需要告诉
gmx pdb2gmx
我们的残差文件中原子的原子和键。我们也可以告诉它角度,但是我们会将它们排除在外,因为
gmx pdb2gmx
会为我们解决这个问题。现在需要在
oplsaa
.
ff
目录中创建以下内容并另存为
methane
.
rtp
:
[ bondedtypes ]
; bonds angles dihedrals impropers all_dihedrals nrexcl HH14 RemoveDih
1 1 3 1 1 3 1 0
; Methane
[ CH4 ]
[ atoms ]
C opls_138 -0.240 1
H1 opls_140 0.060 1
H2 opls_140 0.060 1
H3 opls_140 0.060 1
H4 opls_140 0.060 1
[ bonds ]
C H1
C H2
C H3
C H4
关于上述文件的一些注意事项:[
bondedtypes
]来自
aminoacids
.
rtp
并且是必需的。 在[
atoms
]下的名字可以随便命名,但是需要匹配稍后构建pdb文件中的名字。 注意在第一列中我们给出了原子名称,然后我们给出了原子类型,电荷,然后是电荷组。 在[
bonds
]下,我们只是告诉它每个原子如何连接到其他原子。 在这种情况下,
C
与每个氢都有连接。我们可以选择添加[
angles
],但是如前所述,GROMACS会为我们搞定。 现在关闭文件。 有关这方面的更多信息,请参阅第5.6节。
创建pdb文件并且运行
gmx pdb2gmx
现在我们准备创建pdb文件。 有几个程序可以创建分子结构文件。例如 Avogadro。 另一种方法是使用AmberTools软件包中的“xleap”。 将这个文件保存为methane.pdb。 你的文件应该看起来像这样。 将其保存在您的主目录中的某个位置,但不在
$ GMXLIB
中的任何位置。
在
methane
.
pdb
中将LIG更改为CH4。还要将第一个H更改为H1,将第二个更改为H2等等。PDB文件是固定格式,因此请将每列的开头保留在相同的位置。CONNECT和MASTER记录也不需要,因此可以删除它们。同时继续并将
UNNAMED
更改为
METHANE
。你修改过的文件应该是这样的:
COMPND METHANE
AUTHOR GENERATED BY OPEN BABEL 2.3.2
HETATM 1
C CH4 1 -0.370 0.900 0.000 1.00 0.00 C
HETATM 2 H1 CH4 1 0.700 0.900 0.000 1.00 0.00 H
HETATM 3 H2 CH4 1 -0.727 0.122 0.643 1.00 0.00 H
HETATM 4 H3 CH4 1 -0.727 0.731 -0.995 1.00 0.00 H
HETATM 5 H4 CH4 1 -0.727 1.845 0.351 1.00 0.00 H
END
保存文件为
methane
.
pdb
.
现在我们可以使用
gmx pdb2gmx
创建GROMACS .conf和.top文件:
$ gmx pdb2gmx -f methane
.pdb
系统会提示您选择力场。选择OPLS。如果您在两个不同的力场目录之间有一个选项,请选择您所复制目录中的OPLS。对于水模型选择TIP4PEW。如果您发现GROMACS找不到残基CH4的错误,您可能会使用错误的力场。
将创建三个文件:
conf
.
gro
,
posre
.
itp
和
topol
.
top
。
conf
.
gro
是我们的文件,只包含一个甲烷,
topol
.
top
是系统的拓扑文件,
posre
.
itp
是我们溶质(甲烷)的可选位置限制文件。我们不会使用那个。在
topol
.
top
文件中注意到有一个[
angles
]部分。您还需要在
topol
.
top
中重命名化合物。看看并探索每个文件。 GROMACS手册的第5章将帮助您更多地了解拓扑文件。
注意
:
topol
.
top
和
methane
.
pdb
将在其他教程中再次使用。
对于那些使用
gmx pdb2gmx
生成大型蛋白质拓扑的人来说,事情会变得更加复杂。这仅仅是一个简单的例子,我们可能真的可以在其他地方找到这种拓扑。
溶剂体系
我们的结构文件和拓扑文件到目前为止只有我们的甲烷。我们需要通过使用
gmx solvate
来添加水:
$ gmx solvate -cp conf.gro -o conf.gro -cs tip4p -p topol.top -box 2.3 2.3
2.3
参数文件
我们将使用前一教程中的相同文件。
模拟
我们将使用与上次相同的流程。这假定您的
mdp
文件位于名为
mdp
的目录中:
$ gmx grompp -f mdp/min.mdp -o min -pp min -po min
$ gmx mdrun -deffnm min
$ gmx grompp -f mdp/min2.mdp -o min2 -pp min2 -po min2 -c min -t min
$ gmx mdrun -deffnm min2
$ gmx grompp -f mdp/eql.mdp -o eql -pp eql -po eql -c min2 -t min2
$ gmx mdrun -deffnm eql
$ gmx grompp -f mdp/eql2.mdp -o eql2 -pp eql2 -po eql2 -c eql -t eql
$ gmx mdrun
-deffnm eql2
$ gmx grompp -f mdp/prd.mdp -o prd -pp prd -po prd -c eql2 -t eql2
$ gmx mdrun -deffnm prd
您可以把它当作脚本来使用,在头部添加如下内容:
#!/bin/bash
set -e
然后执行
$ chmod +x run
运行脚本:
$ ./run
其中
set
-
e
表示如果有错误立马停止
分析
我们来计算一下称为径向分布函数的东西。首先,我们需要创建一个索引文件:
$ gmx make_ndx -f conf.gro
> a C
> a OW
> q
然后运行
gmx rdf
$ gmx rdf -f prd.xtc -n index.ndx
在提示时选择
C
作为参考组。 然后选择
OW
。 然后输入
CTRL
-
D
结束。 数据的第1列和第3列中的结果图将通过在gnuplot中执行以下操作来绘制:
> plot 'rdf.xvg' u 1:3 w l
它应该看起来像这样:
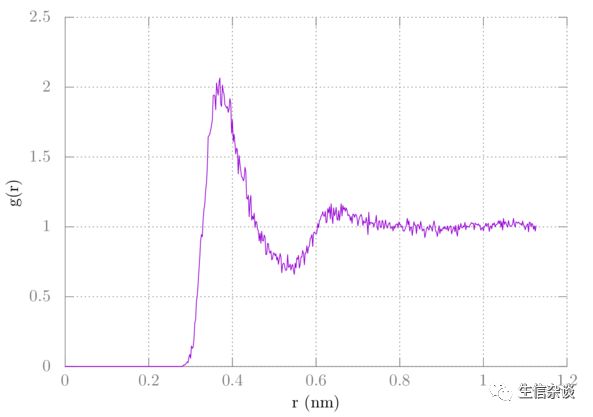
总结
在本教程中,我们学习了如何创建一个用于
gmx pdb2gmx
的残基模板文件(.rtp)。 我们为OPLS甲烷创建了一个结构,并为其生成了一个拓扑。我们用gmx solvated对它周围进行了加水。在此之后,我们进行了仿真,就像上次一样。最后,我们使用
gmx rdf
发现了C-OW径向分布函数。
问题
如果教程中存在错误或错误,或者如果有什么不清楚的地方,请打开一个问题,我很乐意解决它。 如果您有关于GROMACS的一般问题,请参阅GROMACS文档。 您还可以将关于GROMACS的一般问题通过查阅邮件列表来解决。
更多原创精彩视频敬请关注
生信杂谈:






