

摘要
:
目的
研究花椒毒素及其同分异构体对药物代谢酶细胞色素
P450
(
CYPs
)的抑制差异。
方法
采用计算机虚拟技术研究花椒毒素及其同分异构体香柑内酯、异佛手柑内酯与
6
种
CYPs
酶(
CYP1A2
、
CYP2D6
、
CYP3A4
、
CYP2C19
、
CYP2C9
、
CYP2A6
)的相互作用;采用
Amber
(
ff99SB
)软件进行花椒毒素、香柑内酯、异佛手柑内酯与
CYP2A6
的动力学模拟研究;采用
“Cocktail”
药物探针法进行验证,分别测定这
3
种同分异构体对人肝微粒
CYPs
酶活性的影响,以及对
CYP2A6
与
CYP1A2
的半数抑制浓度(
IC
50
)。
结果
计算机模拟技术结果显示,花椒毒素、香柑内酯、异佛手柑内酯对
6
种
CYPs
结合能力不同,对
CYP2A6
的结合能力差异明显。分子对接、分子动力学结果表明,花椒毒素与
CYP2A6
形成的氢键
ASN297∶HD22-M∶O4
是花椒毒素与
CYP2A6
结合过程中最重要的且稳定的氢键,香柑内酯与
ASN297
产生的分子间氢键稳定性低于花椒毒素体系,异佛手柑内酯在动力学中所产生的氢键均不稳定;受体活性口袋中疏水性氨基酸与口袋形状互补是促成配体受体结合的重要因素。
“Cocktail”
药物探针法表明,花椒毒素、香柑内酯、异佛手柑内酯对
CYPs
均具有不同程度的抑制作用,其中对
CYP1A2
具有显著抑制作用,相对剩余酶活力分别为
2.67%
、
4.64%
、
13.58%
,其
IC
50
分别为
1.42
、
2.29
、
5.12 μmol/L
;对
CYP2A6
的抑制作用存在差异,相对剩余酶活力分别为
8.10%
、
38.91%
、
74.98%
,其
IC
50
值分别为
3.89
、
44.03
、
103.30 μmol/L
。
结论
花椒毒素、香柑内酯、异佛手柑内酯对
CYP1A2
具有明显的抑制作用,对
CYP2A6
的抑制效果存在明显差异。甲氧基的取代位置与呋喃环骈合的位置对
3
种化合物与
CYP2A6
结合影响较大,其中
CYP2A6
活性口袋中
ASN297
是蛋白受体与配体结合的关键氨基酸残基。
同分异构体大量存在于中药或天然药物中,研究表明,同分异构体尤其是光学异构体在药理、药动学方面存在很大差异。花椒毒素、香柑内酯、异佛手柑内酯属呋喃香豆素类同分异构体,结构如图
1
所示,这
3
种成分在临床常用中药北沙参、白芷、独活中均有发现
[
1-3
]
,也存在于蔬菜、水果、植物油中
[
4-6
]
。

呋喃香豆素具有广泛的药理活性,在治疗肿瘤、抗炎、抗人体免疫缺陷病毒(
HIV
)方面具有显著优势
[
7-8
]
。现代药理学证明,花椒毒素和香苷内酯具有保护心肌细胞缺氧复氧损伤、抗氧化
[
9
]
、诱导胃癌细胞凋亡
[
10
]
、抗肿瘤的作用
[
11-12
]
。补骨脂素、佛手内酯可抑制淋巴细胞中
HIV-1
复制,具有抗
HIV-1
活性,具有良好的成药前景
[
13
]
。目前研究表明,由呋喃香豆素类化合物引发的药物
-
药物相互作用,主要可概括为对肠道
P-
糖蛋白(
P-gp
)和细胞色素
P450
(
CYP450
,简称
CYPs
)的抑制作用,其中对
CYPs
代谢酶的抑制作用是主要方面
[
14
]
。本实验以
3
种同分异构体对
CYPs
代谢酶抑制活性为标准,借助计算机虚拟技术研究花椒毒素及其同分异构体对
CYPs
的抑制作用差异。
1
材料
1.1
计算机虚拟系统
采用
Discovery Studio
(
DS
,
V4.5
)进行分子对接研究,
Amber
(
ff99SB
)进行分子动力学模拟研究。
1.2
药品与主要试剂
花椒毒素(质量分数
98%
、货号
M1113AS
)、香柑内酯(质量分数
98%
、货号
A0402AS
)、异佛手柑内酯(质量分数
98%
、货号
J1026AS
)、氯唑沙宗(质量分数
98%
、货号
A0206AS
)、奥美拉唑(质量分数
99%
、货号
N0708AS
)、甲苯磺丁脲(质量分数
99%
、货号
S0806AS
),均购自美仑生物科技有限公司;非那西丁(质量分数
98%
,货号
AF20070103
)、卡马西平(质量分数
98%
,货号
AF20070102
)、香豆素(质量分数
98%
,货号
AF20042951
),均购自成都埃法生物科技有限公司;右美沙芬(质量分数
98%
,货号
00367-201305
)、咪达唑仑(质量分数
98%
,货号
171270-201402
)、对乙酰氨基酚(质量分数
98%
,货号
100018-201610
),购自中国食品药品检定研究院;去甲右美沙芬(质量分数
98%
,货号
6700-34-1
,
Sigma
);
1-
羟基咪达唑仑(质量分数
98%
,货号
FD050034-02
,
Cerilliant
);
5-
羟基奥美拉唑(质量分数
98%
,货号
H948863
,
Toronto Research Chemicals
);
4-
羟基甲苯磺丁脲(质量分数
98%
,货号
1-PSB-27-2
,
Toronto Research Chemicals
);
6-
羟基氯唑沙宗(质量分数
98%
,货号
6-QFY-28-2
,
Toronto Research Chemicals
);
7-
羟基香豆素(质量分数
98%
,货号
wkq16072703
,四川省维克奇生物科技有限公司);人肝微粒体(
HLMs
)购于瑞德肝脏疾病研究(上海)有限公司(
SUBK
);
DMSO
、
NADPH
购于美国
Sigma
公司(货号
710N034
、
40836329
);甲醇、乙腈(色谱纯)、甲酸溶液等购于天津康科德科技发展有限公司;所有其他溶剂和化学试剂均为分析纯或以上。
1.3
实验仪器及耗材
十万分之一天平(型号
AX205
,瑞士
MettlerToledo
公司);微型旋涡混合仪(型号
WH-3
,上海沪西分析仪器厂有限公司);高速离心机(型号
ALLEGRA-64R
,美国
Beckman
公司);美国安捷伦公司
6520 Q-TOF LC/MS
,
MassHunter B.02.01SP3
色谱工作站;
Millipore Milli-Q/30L
超纯水机(法国
Millipore
公司);电子恒温水浴锅(天津市泰斯特仪器有限公司);
DKB-501A
型超级恒温水槽(上海精宏实验设备有限公司);海尔
-
86 ℃
超低温保存箱(青岛海尔股份有限公司);
96
、
48
孔培养板(
Corning
公司);微量移液器(
10
、
100
、
200
、
1 000 μL
,
Eppendorf
)。
2
方法
2.1
花椒毒素、香柑内酯、异佛手柑内酯与
CYPs
对接的研究
2.1.1
配体结构的准备
从
PubChem
数据库中下载花椒毒素(
PubChem CID
:
4114
)、香柑内酯(
PubChemCID
:
2355
)、异佛手柑内酯(
PubChem CID
:
68082
)。在
DS
中运行
Minimazizeligand
程序,对配体施加
CharMmforcefield
立场,优化配体小分子。
2.1.2
受体结构准备
从
Protein Data Bank
数据库下载
CYP1A2
(
PDB ID
:
2HI4
)、
CYP2D6
(
PDB ID
:
5TFT
)、
CYP3A4
(
PDB ID
:
4NY4
)、
CYP2C19
(
PDB ID
:
4GQS
)、
CYP2C9
(
PDB ID
:
10G5
)、
CYP2A6
(
PDB ID
:
1Z11
)晶体结构。在
DS
中运行
Prepareprotein
程序优化蛋白结构(加氢、去除无用水分子、补全肽链)。
2.1.3
参数设置
在
DS
中运行半柔性分子对接程序
CDOCKER
,为确保
docking pose
的多样性,设置
Pose Cluster Radius
参数为
0.5 Å
(
1 Å
=
0.1 nm
),其余参数默认。活性位点为靠近
CYP
酶铁卟啉环附近的空腔,符合
PDB
文献记录的位置。
2.1.4
CDOCKER
综合打分一致性评价方法的建立
以往研究中,分子对接多采用系统默认的打分函数
-CDOCKER Energy Score
来评判配体与受体的结合能力。而本研究中花椒毒素、香柑内酯、异佛手柑内酯这
3
种化合物属于同分异构体,分子骨架、电荷分布、分子内能基本相同,仅结构上有细微差别。因此在研究结构相差不大的分子与蛋白作用时,单一采用一种打分函数作为评判标准结果并不可靠。本实验采用多种打分函数对对接结果进行评分,然后对各种函数的结果进行一致性评价,每一种打分的得分越高,一致性评价也就越高。本实验选用了
LigScore1
、
LigScore2
、
-
PLP1
、
-
PLP2
、
Jain
、
-
PMF
、
-
PMF04
、
-
CDOCKEREnergy Score
这几种打分函数对分子对接产生的
10
个构象进综合打分一致性评价。
2.2
花椒毒素、香柑内酯、异佛手柑内酯与
CYP2A6
复合物的动力学模拟研究
取
“2.1”
项分子对接的结果作为动力学模拟的初始构象,分子动力学模拟使用的是
Amber
(
ff99SB
)软件,模拟温度
298 K
,模拟时间
20 ns
。使用最小二乘法拟合蛋白质主链分子的均方根偏差(
RMSD
)值。采用
CPPTRAJ
软件(
https
:
//github.com/Amber-MD/cpptraj
)计算保守氨基酸之间的空间距离。选取最后
4 ns MD
轨迹拍摄的构象用于
MM/PBSA
计算。用
Amber18
计算配体与蛋白质的结合自由能。
Δ
G
值确定根据方程
Δ
G
=
G
complex
-
G
receptor
-
G
ligand
计算得到。
2.3
花椒毒素及其同分异构体对人肝微粒体
CYPs
酶活性的影响
2.3.1
Cocktail
探针药物法的建立
药物对
CYPs
酶活性影响评价研究中,探针底物法是一种广泛的使用方法
[
15
]
。
“Cocktail”
探针药物法是同时给予
2
种或
2
种以上的相对低剂量的探针药物,测定生物样本中每个探针药物的代谢率或者其他代谢类型的指标,以获得多个代谢酶的表型信息。
根据文献报道方法
[
16
]
并加以改进,其中
CYP1A2
的底物为非那西丁(
10 µmol/L
),
CYP2A6
底物为香豆素(
5 µmol/L
),
CYP2D6
底物为右美沙芬(
2.5 µmol/L
),
CYP3A4
底物为咪达唑仑(
5 µmol/L
),
CYP2C9
底物为甲苯磺丁脲(
100 µmol/L
),
CYP2C19
底物为奥美拉唑(
10 µmol/L
)。色谱柱为
Agilent ZORBAX XDB-C
18
(
50 mm
×
2.1 mm
,
3.5 μm
,
SN
:
USHP004037
);进样量
10 µL
;检测波长
280 nm
;体积流量
0.45 mL/min
;离子源参数:
Curtain Gas
为
957.6 Pa
;
IonSpray Voltage 5 000 V
(
ESI
+
)、
-
4 200 V
(
ESI
-
);
Ion Source Gas1
和
Ion Source Gas2
分别为
2.63
、
2.39 kPa
,离子源温度为
350 ℃
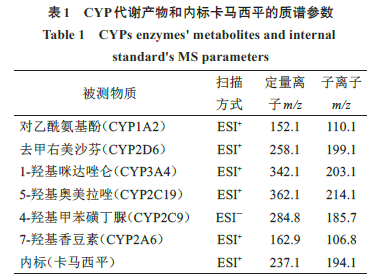
。待测成分和内标的质谱检测参数见表
1
。
2.3.2
人肝微粒体孵育反应 所有的孵育反应都是在37 ℃水浴上进行。预孵育体系的体积为100 µL,包含0.1 mol/L的磷酸盐缓冲液(pH 7.4),2 mg/mL人肝微粒体,花椒毒素、香柑内酯、异佛手柑内酯(终浓度50、5 µmol/L)在加或不加1 mmol/L NADPH(用同等体积的PBS代替)的情况下预孵育30 min后,取20 μL预孵育样品加入到含有100 µL NADPH(1 mmol/L)和80 µL混合探针底物的体系中,孵育15 min,加入400 µL冰甲醇(含内标卡马西平,75 ng/mL)终止反应。4 ℃、10 000×g离心10 min,取上清,HPLC-MS/MS定量分析相应的6种代谢产物。
2.3.3
IC
50
值的测定
实验条件与上述
Cocktail
法相同,不同的是
3
种化合物的终浓度为
0
、
0.3
、
1
、
3
、
10
、
30
、
100
、
300 µmol/L
,探针底物使用非那西丁(
10 µmol/L
)和香豆素(
5 µmol/L
)。通过
GraphPad Prism version 5.0
(
GraphPad software Inc
,
CA
,美国)软件拟合出药物对酶活性的
IC
50
值。
3
结果
3.1
花椒毒素及其同分异构体与
CYPs
的分子对接研究
3.1.1
方法学评估
为确保对接结果的可靠性,需将下载的蛋白活性位点内的配体取下,利用
“2.1”
项中的方法,将配体重新对接进入该活性位点,再对比得分最高的构象与原始构象的
RMSD
值判断该方法是否可以重现配体的原始晶体结构。
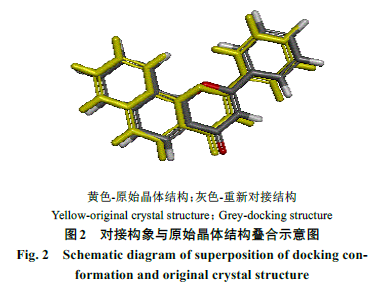
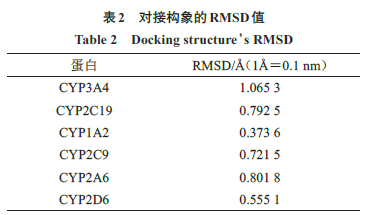
一致性得分最高的构象与原始配体的叠合效果见图
2
,
RMSD
值见表
2
。该方法对
CYP1A2
、
CYP2C9
、
CYP2C19
、
CYP3A4
、
CYP2D6
、
CYP2A6
原始配体的晶体结构均具有良好的重现性,在接下来的实验中将采用这种对接方法。
3.1.2
花椒毒素及其同分异构体与
CYPs
的分子对接得分
采用
“2.1”
项中方法对花椒毒素、香柑内酯、异佛手柑内酯与
CYP1A2
、
CYP2C9
、
CYP2C19
、
CYP3A4
、
CYP2D6
、
CYP2A6
进行了分子对接,其一致性得分如表
3
所示。花椒毒素、香柑内酯、异佛手柑内酯与
CYPs
结合的一致性评价均大于等于
1
,证明在上述的几种打分函数中,至少有一种打分函数认为该化合物能与蛋白产生结合。
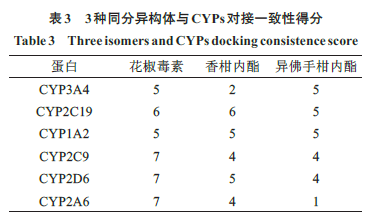
3
个同分异构体与
CYP2A6
对接的一致性评分分别为
7
、
4
、
1
,差异明显。为了研究
3
个同分异构体与
CYP2A6
结合方式的差异,表
4
中列出了
3
种化合物与
CYP2A6
的结合方式。图
3
所示
3
个同分异构
体在
CYP2A6
活性口袋中的构象与
2D
图。对接结果显示,花椒毒素能与
CYP2A6
活性中心的
ASN297
产生一个常规氢键,键长
1.88 Å
,两个碳氢键,键长分别为
2.24
、
2.41 Å
;香柑内酯不能与
CYP2A6
产生常规氢键,只能与
CYP2A6
活性中心的
ASN297
产生一个碳氢键,键长
2.36Å
;异佛手柑内酯不能与
CYP2A6
产生任何分子间氢键。

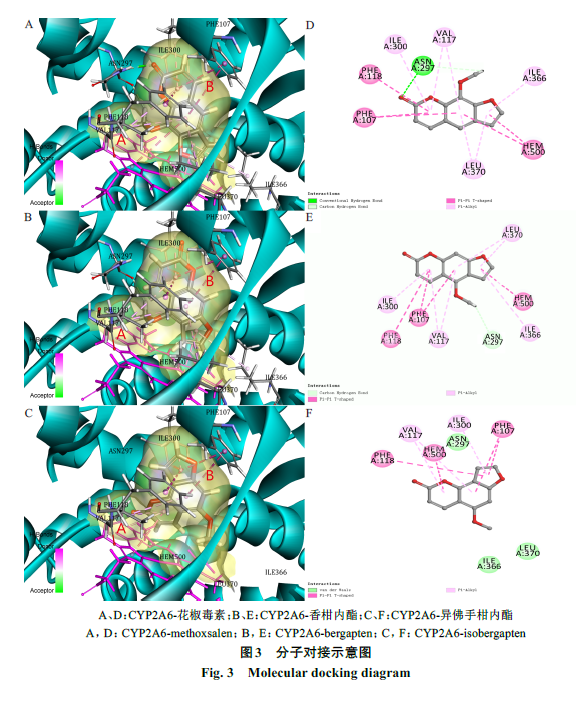
除此之外,
CYP2A6
活性口袋中有
2
个
PHE
(
PHE117
、
PHE118
)和辅酶
HEM500
,这些分子中都具有环状结构会与苯环等环状结构形成
Pi-Pi Tshaped
相互作用。这
3
个同分异构体均具有苯骈
α
吡喃酮结构,能与活性口袋发生
Pi-Pi T shaped
相互作用,从而占据活性口袋。为进一步考察对接体系与分子间氢键的的稳定性,本实验对
3
个对接体系进行了分子动力学模拟。
3.2
花椒毒素及其同分异构体与
CYP2A6
结合的分子动力学模拟研究
3.2.1
分子动力学模拟稳定性的探究
分别对受体
CYP2A6
体系和所有的蛋白
-
配体复合物体系
CYP2A6-
花椒毒素、
CYP2A6-
香柑内酯、
CYP2A6-
异佛手柑内酯进行
20 ns
的分子动力学模拟。为了确定分子动力学模拟结果的稳定性,在各个体系中均以其初始蛋白构象为参照,在模拟时间内对系统
Cα
原子的
RMSD
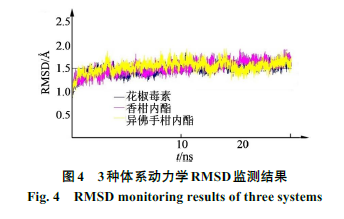
进行计算。结果如图
4
所示,
CYP2A6-
花椒毒素、
CYP2A6-
香柑内酯、
CYP2A6-
异佛手柑内酯的平均
RMSD
值为
1.52
、
1.47
、
1.53 Å
,证明动力学动态模拟轨迹平衡性是可靠的,可用于后续进一步分析。
3.2.2
各体系分子间氢键稳定性分析
复合物体系中配体与受体之间的氢键稳定性可以反映出配体与受体之间相互作用的强弱,针对
3
种复合物体系进行氢键稳定性分析,结果见表
5
。在整个动力学模拟过程中,共采样
20 000 ps
数据。花椒毒素与
CYP2A6
形成的氢键
ASN297∶HD22-M∶O4
出现
12 909 ps
,占有率为
64.54%
,平均距离为
2.84 Å
,平均角度为
154.93°
,是花椒毒素与
CYP2A6
结合过程中最重要的且稳定的氢键。香柑内酯与
ASN297
产生的分子间氢键在动力学中共出现
10 122 ps
,占有率为
50.61%
,低于花椒毒素体系,异佛手柑内酯在动力学中所产生的氢键均不稳定。

3.2.3
各体系结合自由能的计算
蛋白和小分子的结合自由能可以从能量的角度表征受体与配体之间键合的能力。结合能负值越大,表明键合能力越强,破坏该键所需能量也就越大。选取
3
个体系最后
4 ns
进行相对结合自由能计算,结果见表
6
。
3
个同分异构体化合物与
CYP2A6
之间的结合自由能均为负值,说明
3
种化合物均能与
CYP2A6
产生结合。将结合自由能进行分解,范德华相互作用(
G
vdw
)、静电相互作用(
G
ele
)和非极性溶剂化自由能(
G
esurf
)均为负值,说明
G
vdw
、
G
ele
、
G
esurf
这
3
种相互作用能够诱导配体受体结合,而极性溶剂化自由能(
G
egb
)为正值说明其不利于配体分子结合到受体蛋白中。从表中还可知,
G
vdw
对配体受体结合最为显著,由此可以判断,受体活性口袋中疏水性氨基酸与口袋形状互补是促成配体受体结合的重要因素。
3.3
花椒毒素及其同分异构体对
CYPs
酶的抑制
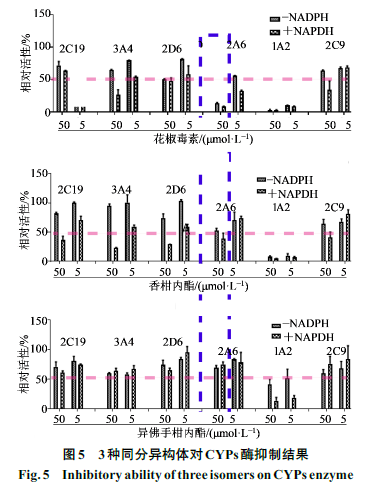
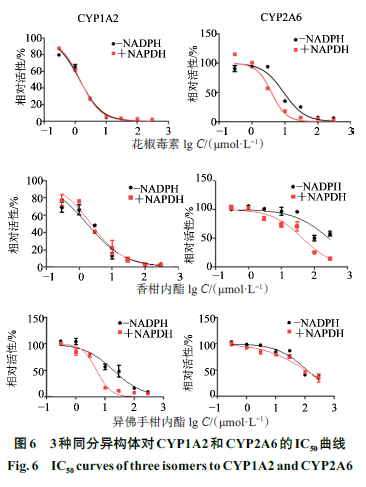
如图
5
、
6
所示,花椒毒素、香柑内酯、异佛手柑内酯对
CYPs
均有不同程度的抑制,在
5
和
50 μmol/L
时均对
CYP1A2
显示出非常明显的抑制作用。在
NADPH
存在下,花椒毒素、香柑内酯、异佛手柑内酯浓度为
50 μmol/L
时,
CYP1A2
的相对剩余酶活力分别为
2.67%
、
4.64%
、
13.58%
,
IC
50
值分别为
1.419
、
2.289
、
5.124 μmol/L
,表明
3
个同分异构体对
CYP1A2
均有较强的抑制作用;
CYP2A6
的相对剩余酶活力分别为
8.10%
、
38.91%
、
74.98%
,
IC
50
值分别为
3.893
、
44.03
、
103.30 μmol/L
。体外酶的抑制实验,进一步验证了分子对接和分子动力学结果的可靠性。
4
讨论
4.1
花椒毒素及其同分异构体构效关系的探讨
本实验使用的是半柔性分子对接方式,在分子对接过程中固定了受体活性口袋的形状,更有利于分析因配体的结构不同而导致作用方式差异的原因。从表
4
中可以看出,
3
个同分异构体均能与
CYP2A6
活性口袋中的
PHE118
、
PHE107
、
HEM500
产生
Pi-Pi T shaped
相互作用,与
VAL117
、
ILE300
产生
Pi-Alkyl
相互作用。因此不难看出,
3
个同分异构体与
CYP2A6
结合产生差异的主要原因是分子间氢键的作用。
从图
3
可以看出,甲氧基与呋喃环取代的位置不同和活性口袋的空间特征是造成差异的主要原因。
CYP2A6
活性口袋切面大致成三角形,
A
侧空间较大而
B
侧空间较小。对于线型分子花椒毒素与香柑内酯,由于
CYP2A6
口袋中
B
侧没有足够的空间,使得甲氧基只能存在于
A
侧。花椒毒素上的甲氧基属于
8
位取代,与羰基在同侧;香柑内酯上的甲氧基属于
5
位取代,与羰基在异侧,所以花椒毒素的羰基会靠近
ASN297
形成氢键,香柑内酯的羰基远离
ASN297
不会形成分子间氢键。对于异佛手柑内酯,其
7
,
8
位骈合呋喃环属于角型分子,呋喃环、吡喃酮环、甲氧基在空间位置上组成三角形结构,由于呋喃环小、吡喃酮环与甲氧基形成的角度较大,所以呋喃环进入
B
侧较小的空间,吡喃酮环与甲氧基在口袋
A
侧,与此同时
PHE117
、
PHE118
与结构中吡喃酮环产生
Pi-Pi T shaped
相互作用最终导致吡喃酮环在左侧,甲氧基在右侧。正是由于这种几何特征导致异佛手柑内酯不会与口袋中任何氨基酸残基形成稳定的分子间氢键,这是其结合能力较差,对
CYP2A6
的抑制能力低的主要原因。
分子动力学模拟充分模拟了人体内生理环境,计算过程考虑到受体蛋白的柔性。在考虑到蛋白柔性后,发现香柑内酯在动力学模拟过程中也可以与
ASN297
产生氢键作用但稳定性低于花椒毒素与
CYP2A6
产生的氢键,异佛手柑内酯则不会产生稳定的分子间氢键。在动力学模拟中,
3
种同分异构体与分子对接构象大致类似,对于线型分子花椒毒素、香柑内酯,甲氧基均在
A
口袋,角型分子香柑内酯的吡喃酮环均在
A
口袋,进一步阐明了分子对接的结果。通过氢键稳定性分析与计算相互作用能,推测出
CYP2A6
口袋中疏水性氨基酸与
ASN297
为关键氨基酸残基,与分子对接结果相同。从上述实验中不难看出,呋喃环的骈合位置与甲氧基的取代位点是造成这
3
种同分异构体活性差异的根本原因。
6
,
7
位骈合呋喃环(花椒毒素、香柑内酯)更容易与
CYP2A6
的活性口袋产生稳定结合,当
7
,
8
位骈合呋喃环(异佛手柑内酯)将大大降低化合物与
CYP2A6
的结合能力;当
5
位引入甲氧基时(香柑内酯),由于空间效应的影响,减弱化合物与
CYP2A6
的结合能力。
宋志英等
[
16
]
研究了花椒毒素及其同分异构体与溶菌酶
LYSO
的结合机制,结果同样表明呋喃环和甲氧基位置的不同导致了
4
种呋喃香豆素类药物与
LYSO
的作用强弱不同,其作用力顺序依次为
8-
甲氧基补骨脂素(花椒毒素)>
5-
甲氧基补骨脂素(香柑内酯)>异补骨脂素(异佛手柑内酯)>补骨脂素。不难发现,在呋喃香豆素中,呋喃环的骈合位置与
5
,
8
位取代将对其药理活性具有较大影响。
4.2
花椒毒素及其同分异构体对药物代谢的抑制作用
花椒毒素及其同分异构体具有广泛的药理活性和良好成药前景,因此研究这
3
个同分异构体对
CYPs
的影响是评估药物相互作用和临床不良反应的重要组成部分
[
17
]
。肝微粒体
CYPs
具有广泛的底物特异性,是药物与外源性物质
Ⅰ
相代谢的重要场所,约有
60%
的处方药经
CYPs
酶代谢,是参与药物代谢的主要酶,在药物代谢酶中起至关重要的作用
[
18
]
。由于药物对
CYPs
的诱导或抑制而产生的药物
-
药物相互作用(
DDI
)是多种药物被撤市的主要原因。其中
CYPs
抑制剂所导致
DDI
的临床意义远大于酶诱导作用。有研究发现,
CYPs
的抑制剂可增加体内经
CYPs
代谢的药物浓度,使药物代谢减慢、作用时间增长,从而延长药物在体内处置时间
[
19-20
]
。因此,
CYPs
酶的活性将会直接影响经其代谢药物的药动学从而增加药物在使用过程中产生不良反应的可能,引起严重的
DDI
,影响药物治疗的安全性和有效性。
“Cocktail”
实验结果表明,
3
种化合物均对
CYP1A2
产生明显的抑制作用。
CYP1A2
是一种重要的
CYPs
,主要分布在肝脏中,其含量排名第
3
[
21
]
。
CYP1A2
参与咖啡因、普萘洛尔、硝苯地平等
20
多种药物的代谢过程,同时也负责一些内源性激素如褪黑激素、胆红素、雌二醇的羟化反应
[
20
]
。因此含有这
3
种成分的药物和食物应避免与主要经
CYP1A2
代谢的药物同时服用而发生相互作用或不良反应。
CYP2A6
在尼古丁氧化代谢中起重要作用,主要参与甲硝唑、替加氟、来曲唑等临床常用药物的代谢。研究表明,
CYP2A6
是人肝微粒体中最主要、可能也是唯一参与香豆素
7-
羟化反应的酶
[
22
]
。因此含有花椒毒素的药物在临床使用中应尽量避免与含有香豆素、经
CYP2A6
代谢的药物同时使用产生
DDI
,为临床联合用药提供参考。
4.3
计算机虚拟技术在构效关系研究的展望
药物的生物学活性及代谢特征与其分子结构密切相关,绝大多数药物通过与体内靶点结合从而产生药效。在结合过程中,需要药物分子和体内靶点满足几何匹配与能量匹配,由于小分子药物和靶点蛋白具有一定的柔性,因此在两者相互作用过程中契合程度越高的体系往往特异性和生物活性越强。同分异构体化合物,虽然具有相同的分子式,大致相同的母核结构,但是由于取代基位置的不同,在很大程度上将影响其与靶点蛋白的契合程度。特别是具有手性中心的化合物,当其对映体与受体分子满足契合具有较强的生物活性,那么另一对映体必然因为不能满足契合从而活性大大降低。例如氧氟沙星、吲哚美辛均为
S
异构体活性分子,而
R
异构体没有活性
[
23
]
。
在以往的构效关系研究中
[
24-26
]
,多采用生物学实验结合光色谱的研究方法,前者通过生物学实验获得目标分子的生物学性质;后者通过分析的方法获得目标分子的结构特征,综合前两部分实验结果进行论述与猜测。而计算机虚拟技术的应用更加直观地描述药物结构与受体的契合关系,能直接
“
看到
”
分子结构上的差异是如何影响其与靶点的作用方式,也可直接清楚阐明药物的结构特征与其生物学活性关系,即定量构效关系(
Q-SAR
),并且能对即将开展的生物实验进行指导。
花椒毒素及其同分异构体香柑内酯、异佛手柑内酯对
CYP1A2
具有明显的抑制作用,对
CYP2A6
的抑制效果存在明显差异。其中,甲氧基的取代位置与呋喃环骈合的位置对
3
种化合物与
CYP2A6
结合能力具有较大影响,
CYP2A6
活性口袋中
ASN297
与花椒毒素
2
位羰基在整个动力学过程中表现非常稳定,是蛋白受体与配体结合的关键氨基酸残基。
利益冲突
所有作者均声明不存在利益冲突
参考文献(略)
来 源:刘世豪,王丽莉,刘文莉,靳超群,陈佳萍,何新. 基于计算机虚拟技术研究花椒毒素及其同分异构体对细胞色素P450的抑制作用差异 [J]. 药物评价研究, 2021, 44(1):15-24.







