[作者简介] 1990 - 1995 年就读于沈阳药科大学日语药学专业,1995 - 1998 年就读于沈阳药科大学药物分析专业硕士研究生。毕业后在上海市食品药品检验所工作至今,深谙行业现状。2003 年 8 月 - 2004 年 2 月赴日本国家药品检验所进修,回国后始终致力于国产仿制药的品质提升。
自本人大力呼吁和宣传:众人切勿仅关注美国 FDA 网站,日本药品审评中心 (PMDA) 网站资料也是“价值连城”、尤每一产品的《研发申报资料概述(简称日本IF文件)》后,收到多位同仁来电来函,纷纷表示:没想到日本 PMDA 上的资料公开得如此详尽,比美国 FDA 还多,有的企业公开内容甚至相当于《半套资料》,对我们正在进行的该品种仿制药研发极具借鉴意义和帮助作用。您在日本国家药检所进修学习过,知晓日本官方这么做的出发点吗?

这一疑问引发了本次撰文,因在下深知:PMDA 的这一做法绝非是“冒傻气”,而是为了追求公正公平的审评,以排除各企业间的相互猜忌,求得利益分配的合情合理与心平气和,最终构建和谐美满的行业氛围与竞争环境。详述如下:
1.日本厚生省(负责医疗卫生和社会保障的主管部门)要求 PMDA 在保证仿制药质量不劣于原研药的前提下、尽可能多批准
厚生省秉承节省医保开支和降低国民用药负担的出发点,要求 PMDA 在保证仿制药质量等同于原研药的前提下,尽可能地多批准(美国政府对 FDA 的要求亦如此)。
为此,PMDA 绝不会蛮不讲理地提高技术门槛,批准家数完全依照“原研药自身含金量”以及“市场容量与自我调节”来确定。如苯磺酸氨氯地平片,由于原研药的制剂难度不大,仿制药研发易如反掌,使得大量企业申报,PMDA “来者不拒”,截止目前共批准了近 200 家生产企业文号。
同时,发达国家多年前就放弃了原料药生产,均是从我国和印度的原料药厂或化工厂直接采购,故这些国家的仿制药企业已将全部精力放在制剂开发、尤工业药剂学大生产上,使得每一企业的制剂能力大幅提升。
国家这一导向也极大地促进了中小企业发展,并对社会就业带来积极影响。
2. 日本的《立卷审查》——PMDA 网站公开的《IF 文件》内容
为做到公平公正的审评,PMDA 自 2004 年 4 月 1 日成立之初,就按照《国家机关相关信息公开法》树立了“尽可能公开”的审评原则与工作宗旨。因唯有公开,才能杜绝审评员暗箱操作和内部的腐败滋生。
为此,任何一个产品的《IF 文件》均被要求公开,这就是日本的《立卷审查》,且不惧怕竞争对手抄袭。公开的主要内容如下(针对“小分子化学药口服固体制剂”):
△ 一般性药学研究
△ 杂质研究(基本无,呵呵~~。因制剂研究仅需关注“原料药制成 0 天制剂”和“ 0 天制剂在货架期”这两个环节产生的杂质,而很多产品均十分稳定)
△ 制剂尺寸、形状与片重
△ 辅料种类
△ 稳定性考核:加速试验和长期试验 6 个月各项考察指标检测结果(均为具体数据、如有多种包装,均需罗列上)
△ 多条溶出曲线比对研究的详尽信息:试验方法、测定数据,比较结果等
△ 生物等效性 (BE) 试验所有数据和结果
△ 其他大量内容(请自我下载学习)
△ 辅料含量和制剂工艺(唯有该内容属于核心机密、不能公开)

每一药品按规格分别罗列参比制剂与仿制制剂,同时公布每家企业产品的单位药剂价格、1 日最大服用量药价和最小服用量药价,因该国处方药销售实行“国家专卖制度”——在全国任何一家医院或药房的价格均是一致的、公开的。
其中,所有仿制药售价都一致(随家数不断增多、国家会不断下调价格),唯有参比制剂售价稍高一些,这些信息使查阅者一目了然、清清楚楚。
更令人称奇的是:还公开每家仿制药企业 BE 试验时的受试者例数、投药量、AUC、Cmax、Tmax、T1/2 等具体数据。
举例如下:截止目前,PMDA 共批准了 20 余家阿托伐他汀钙片仿制药,每家企业均有 5mg 和 10mg。大多数企业均是遵循“尽力做到体外溶出行为与原研药一致,其后 BE 试验水到渠成”的研发路径。
此时,BE 试验受试者便可采用通常的 18 - 24 例年轻男性。如“日本仿制药株式会社”研制的产品:按国家规定,两规格各与相同规格的原研药比较了 5 条曲线,结果除 6.8 介质中的溶出行为均不一致外,另外 8 条曲线均一致,此时两规格的 BE 试验都可采用通常的24例受试者。
【注】针对普通制剂,在相同试验条件下、如原研药在 6.8 介质中的溶出行为明显慢于自身其他介质溶出行为时,允许在开发仿制制剂时,将 6.8 介质中的溶出行为做得比原研药释放得更充分、更彻底些,以满足药物在胃酸缺乏患者(通常为中老年患者,体内易呈现高 pH 值)体内生物利用度依然良好的效果,这种研发方式属于“仿制药创新”。具体如图(空圈为仿制药、实圈为原研药):
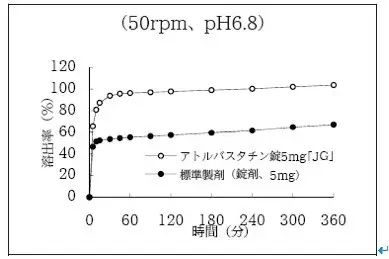
而另一家企业“日本明治制药株式会社”,比较的 10 条溶出曲线中有 6 条不一致,尤关键的 1.2 介质、5.0 介质和水介质均不一致(在这些介质中、原研制剂呈现缓慢释放情形,就是本人常说的“那根筋”)。
但该企业坚持认为“仿制药质量等同于原研药的判断不是依靠体外溶出行为一致、而是以体内生物利用度一致为准”,提出开展BE试验申请。
此时,PMDA 会允许挑战,但必须在受试者种类(如选用胃酸缺乏人群)或受试者例数(很多企业采取此法)上“补回来”,最终该企业直至做到 60 例(5mg)和 120 例(10mg)才通过。值得庆幸的是、最终还是通过了,否则将“竹篮打水一场空”。
PMDA 如此要求的出发点是:企业必须在溶出与 BE 上“靠一头”:体外溶出行为的一致性代表该公司在制剂研发上的精益求精、孜孜以求,想必付出了很大代价,所以 BE 试验可轻松些。
反之、则说明该企业的制剂研发尚不够深入、制剂人员水平仍有待提升,此时如企业强求实施 BE 试验,就必须在该实验上有所弥补和改变,以平衡各企业间研发经费付出与利益分配。如出现“一头也靠不上”情形,欢迎“检举揭发”。如此,当以上两家企业研发总监见面时就会“相逢一笑泯恩仇”了。
这就是我们在查阅该网站时会惊异发现:同一品种的多家仿制药企业 BE 试验受试者例数会呈现 9 - 30 例或 24 - 120 例巨大波动的原因。PMDA 的工作原则就是通过公开方式,让各获批企业间心悦诚服,否则就会出现“相互怀疑、不服举报”的极端案例发生。
还记得在日进修时的一个场景:门厅处有 2 个像电话亭似的小格子间,是专为对国家经费使用抱有疑问、前来查询的公民准备的。也就是说:任何一个纳税人,如对国家机构使用的经费提出质疑,都欢迎前来查询财务账目!
对国家机关和事业单位建立这样的行政要求,我们就不难理解为何 PMDA 会“傻乎乎”地公开各家企业的研发资料了;同时,我们也不必有“ PMDA 察觉到近期来自中国的查询和下载特别多,将屏蔽掉中国网民”的顾虑。
小结:
药物研发对于企业而言具有投资大、风险高的特性,且国家审评结论与审批结果对市场瓜分和利益分配影响巨大。为杜绝审评中出现偏颇与腐败,日本这种公开《每家企业申报材料》的方式是否值得我国药审中心 (CDE) 借鉴与效仿呢?
CDE 目前已有工作人员 600人,2020 年将增至 1600 人,可谓兵强马壮、人多势众。本人相信:在人手如此充足的情况下、如欲效仿 PMDA 作法完全可实现。衷心期望本文能为 CDE 的发展与进步提供样本和素材。
【声明】本文仅代表个人观点,不代表所在单位。
本文转载至微信公众号「健言」
【延伸阅读】
1. 【终极解密】BCSII 和 IV 类普通制剂关键性溶出曲线
2. 谢沐风:以科学的溶出度方法「迎战」一致性评价和制剂国际化(美国与日本仿制药审评模式的碰撞与融合)
3. 【经世致用 VS 过度理论化】 谢沐风:对「体外不一致、体内一致」案例的解读
4. 制剂真的好难|谢沐风老师与网友的精彩互动
5. 对「FDA 批准的中国仿制药:理想 PK 现实」一文有感

点击「阅读原文」,即可免费获取一个 Insight 账号,查阅更多 PMDA 公开资料










