大部分情况下,从CRAN或Bioconductor上安装R包并不是一件复杂的事情。
但有一类问题非常让我难受,那就是明明已经安装的R包,却在加载的时候告诉我用不了。
> library("clusterProfiler")
错误: package or namespace load failed for ‘clusterProfiler’ in loadNamespace(j i[[1L]], c(lib.loc, .libPaths()), versionCheck = vI[[j]]):
不存在叫‘DO.db’这个名字的程辑包
于是,我就得复制DO.db,然后用
BiocManager
::
install
(
"DO.db"
)
安装那个R包。运气好的话,安装完一个R包就行,但是运行不好,你可能就要连环安装多个包。
这个问题只会在部分电脑上出现,估计是包的依赖解析出现了问题,你可能无法通过重装之前的R包解决,就得一直复制粘贴,所以让我非常暴躁。当然我遇的多了,顶多是复制粘贴几次就能搞定,但是对于小白而言,估计就束手无策了。
为了搞定让自己一劳永逸的搞定问题,我就写了一个fxxkit函数,专门搞定这问题
通过下面这行代码加载我的函数,
source("https://raw.githubusercontent.com/xuzhougeng/myscripts/master/zgtools.R")
然后当下次遇到问题时,勇敢的fxxkit
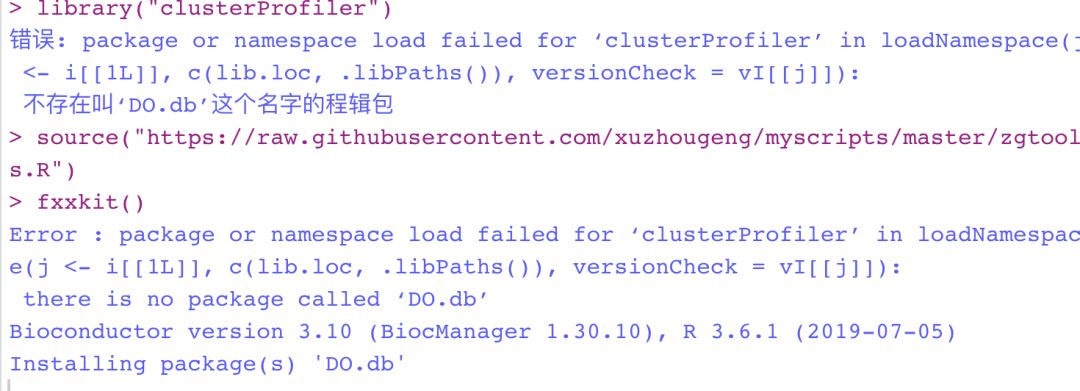
如果你发现自己缺少一个包(注意,这个包必须得有哦),JUST fxxkit()

最终你的R包装好了,也达到了inner peace。
另附源代码
fxxkit function(){
# set the language to english
env Sys.getenv("LANGUAGE")
on.exit(Sys.setenv("LANGUAGE" = env))
Sys.setenv("LANGUAGE" = "en")
# get the history
file1 tempfile("Rrawhist")
savehistory(file1)
rawhist readLines(file1)
unlink(file1)
# get the package name
rawhist rawhist[grepl("^library|^require", rawhist)]
rawhist rawhist[length(rawhist
)]
package.name strsplit(rawhist, "\\(|\\)")[[1]][2]
package.name gsub("'|\"","", package.name)
# get the dependent package
text try(library(package.name, character.only = TRUE))
# extract the package name from error message
package.name gsub(".*called ‘(.*?)’.*", "\\1", text[1])
# download package
if (!requireNamespace("BiocManager", quietly = TRUE)){
install.packages("BiocManager")
}
BiocManager::install(package.name)
}




