照例回顾
基因组学—-基因家族分析(一)
基因组学—-GO注释
基因组学—-启动子预测
基因组学—-基因组的拼接实例
为什么要提取intron位置
有些时候为了分析某些非编码RNA的表达,需要以intron区域的表达做为对照;
有些时候需要分析某些基因组元件是否位于基因组intron区域;
有些时候需要分析不同物种intron的多少、长短以及所占基因组的比例;
有些时候就是单纯想看一下基因组的每个基因有多少intron;
好了,这就需要我们自己编写程序拿到intron的位置,然后再进行进一步的计算!如果我懒的写程序或者不会写呢?
今天就给大家介绍一种简单点的方法
方法一
软件 genometools
下载地址
http://genometools.org/pub/
安装
make -j4 install prefix=~/gt
prefix 后面的路径就是需要安装的目录
我们安装的其实是个软件包,里面功能很强大;有时间的小伙伴可以仔细研究一下它的种种功能,如有机会我们会继续介绍!
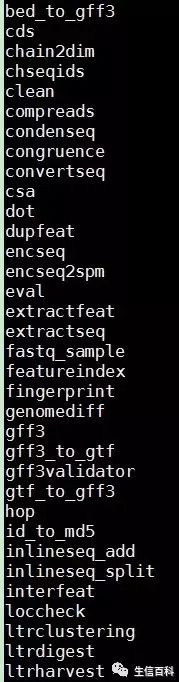
譬如:
-
bed 转换为gff 文件
-
gff3 转换为 gtf 文件;
-
预测 基因组的 LTR 类型转座子;
-
对LTR转座子进行聚类;
-
根据位置提取基因组序列;
-
翻译cds序列;
-
寻找ORF;
-
分割fasta序列;
。
。
。
是不是很强大!

用法
gt gff3 --addintrons Arabidopsis.gff >Arabidopsis.add.gff &
其中
Arabidopsis.gff 就是所有数据库中都可以下载到的基因位置文件;
运行之后,intron就可以当成一个feature加到该文件中。
方法二
给个python程序吧,如果程序报错,自己稍微修改一下哦!
程序名字:
extract_intron_gff3_from_gff3.py
# Insert 'intron' entries to GFF.
import time
import os
import misopy
import misopy.gff_utils as gff_utils
import misopy.Gene as gene_utils
def instert_introns_to_gff3(gff_filename, output_gff3_filename):
output_filename = os.path.join("%s_introns.gff3" %(output_gff3_filename))
print "Adding introns to GFF..."
print " - Input: %s" %(gff_filename)
print " - Output: %s" %(output_filename)
gff_out = gff_utils.Writer(open(output_filename, "w"))
gff_db = gff_utils.GFFDatabase(from_filename=gff_filename, reverse_recs=True)
t1 = time.time()
genes = gene_utils.load_genes_from_gff(gff_filename)
for gene_id in genes:
gene_info = genes[gene_id]
gene_tree = gene_info["hierarchy"]
gene_obj = gene_info["gene_object"]
gene_rec = gene_tree[gene_id]["gene"]
gene_start = int(str(gene_tree[gene_id]['gene']).split(",")[3].strip(" "))
gene_end = int(str(gene_tree[gene_id]['gene']).split(",")[4].strip(" "))
# Write the GFF record
gff_out.write(gene_rec)
# Write out the mRNAs, their exons, and then
# input the introns
for mRNA_id in gene_tree[gene_id]["mRNAs"]:
curr_mRNA = gene_tree[gene_id]["mRNAs"][mRNA_id]
gff_out.write(curr_mRNA["record"])
# Write out the exons
curr_exons = gene_tree[gene_id]["mRNAs"][mRNA_id]["exons"]
#curr_cds= gene_tree[gene_id]["mRNAs"][mRNA_id]["CDSs"]
for exon in curr_exons:
gff_out.write(curr_exons[exon]["record"])
#gff_out.write(curr_cds[cds]["record"])
# Now output the introns
for isoform in gene_obj.isoforms:
#print gene_obj.isoforms
intron_coords = []
for first_exon, second_exon in zip(isoform.parts,
isoform.parts):
final_exon_end=0
first_exon_start=0
if(len(intron_coords)==len(isoform.parts)-1):
final_exon_end=second_exon.end
if(len(isoform.parts)==1):
first_exon_start=first_exon.start
#print first_exon
if(first_exon_start>1 and gene_start==1):
intron_start=1
intron_end = first_exon_start - 1
elif(gene_end>final_exon_end and final_exon_end!=0):
#print str(isoform.parts[len(isoform.parts)-1]).split(",")
print first_exon_start
intron_start=final_exon_end+1
intron_end=gene_end
else:
intron_start = first_exon.end + 1
intron_end = second_exon.start - 1
if intron_start >= intron_end:
continue
intron_coords.append((intron_start, intron_end))
# Create record for this intron
intron_id = "%s:%d-%d:%s" %(gene_obj.chrom,
intron_start,
intron_end,gene_obj.strand)
intron_rec = \
gff_utils.GFF(gene_obj.chrom, gene_rec.source, "intron",
intron_start, intron_end,".",gene_obj.strand,
attributes={"ID": [gene_obj.label],
"Parent": [isoform.label]})
gff_out.write(intron_rec)
for first_exon, second_exon in zip(isoform.parts,
isoform.parts[1::1]):
# Intron start coordinate is the coordinate right after
# the end of the first exon, intron end coordinate is the
# coordinate just before the beginning of the second exon
#print "test"
#print "s"
#else:
# intron_start = first_exon.end + 1
# intron_end = second_exon.start - 1
intron_start = first_exon.end + 1
intron_end = second_exon.start - 1
if intron_start >= intron_end:
continue
intron_coords.append((intron_start, intron_end))
# Create record for this intron
intron_id = "%s:%d-%d:%s" %(gene_obj.chrom,
intron_start,
intron_end,gene_obj.strand)
intron_rec = \
gff_utils.GFF(gene_obj.chrom, gene_rec.source, "intron",
intron_start, intron_end,".",gene_obj.strand,
attributes={"ID": [gene_obj.label],
"Parent": [isoform.label]})
gff_out.write(intron_rec)
t2 = time.time()
print "Addition took %.2f minutes." %((t2 - t1)/60.)
if __name__=="__main__":
import sys
if len(sys.argv) > 1:
file = sys.argv[1]
store = sys.argv[2]
instert_introns_to_gff3(file, store)
else:
sys.exit("No input")
怎么用呢?
python extract_intron_gff3_from_gff3.py [input.gff3] [output.gff3]

对了,生信资料赠送依然有效,欢迎大家积极参与 ! 阅读原文参与!
欢迎分享!