近年来研究表明,LncRNA参与了多种重要的调控过程,如X染色体沉默、基因组印记及染色质修饰等。通过LncRNA测序,可以快速获得与其生物学过程或者疾病相关的LncRNA的表达变化,从而促进LncRNA的深入研究。今天,小编就为您介绍一款精准、快速且造价较低的高吞吐量非编码RNA测绘方法。

该方法被称为“RNA捕获长测序(RNA Capture Long Seq,CLS)”,它偶联靶RNA捕获和Pacific
Biosciences(PacBio)三代长读长测序,采用最先进的测序方法进行扩增和分析,专门针对基因组的非编码区,可用于改进已登记的基因模式或鉴定新基因位点。
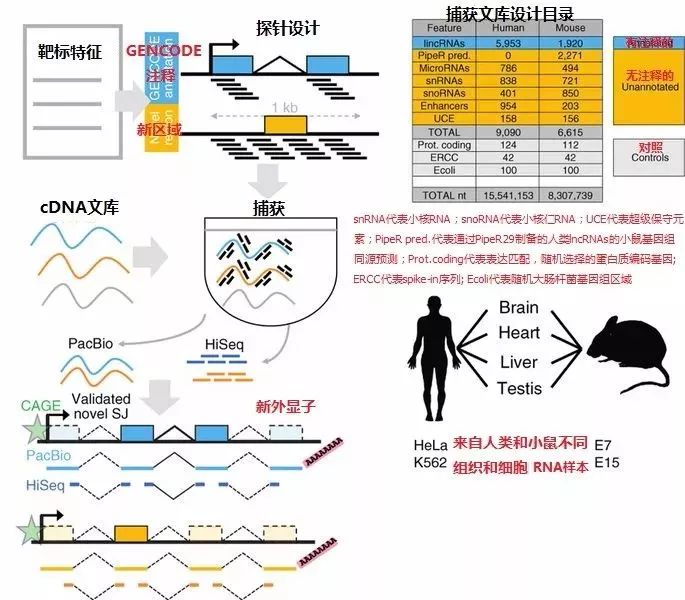
(自动化、高质量转录组注释战略)
用CLS捕获有注释的(蓝色)或绘制可疑位点(金色)的新转录组结构(下方黑色短线代表寡核苷酸,用来捕获跨区目标)。其中,PacBio
文库直接来自被捕获分子,而Illumina HiSeq短读长测序则用于预测剪接点(splice junctions,SJ)的独立确认。使用CAGE簇(CAGE
clusters)确认预测转录起始位点,PacBio读取(红色)通过非基因组编码polyA+测序确认转录终止位点。浅蓝色的虚线轮廓矩形代表新外显子。
snRNA代表小核RNA;snoRNA代表小核仁RNA;UCE代表超级保守元素;PipeR
pred.代表通过PipeR29制备的人类lncRNAs的小鼠基因组同源预测;Prot.coding代表表达匹配,随机选择的蛋白质编码基因;
ERCC代表spike-in序列; Ecoli代表随机大肠杆菌基因组区域(增强子和UCEs的两股链都设计了探针,在这里分开计算)。
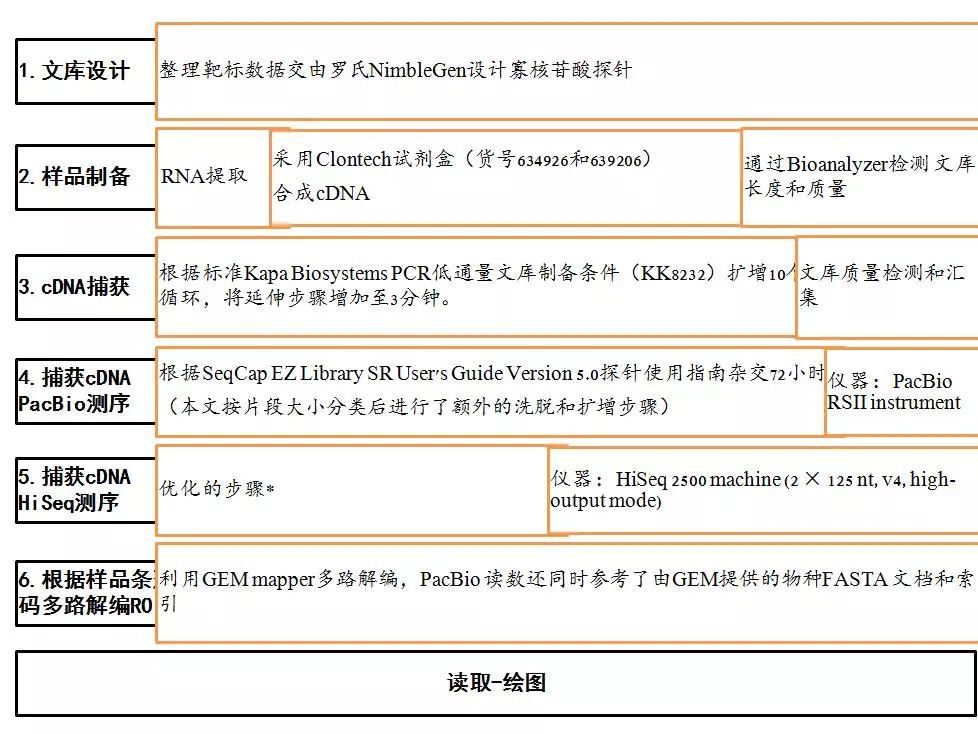

(技术路线)
研究人员将被捕获的cDNA按大小分为三组:1-1.5kb、1.5-2.5kb和2.5kb以上,随后被用以构建PacBio
单分子实时(SMRT)测序技术的测序文库。
130个SMRT细胞样品测序总计获得人类和小鼠各获得200万个读长。通过多路解编PacBio读取(ROIs)以及全基因组绘图,结果显示出了非常高的绘图率(两种物种都大于99%),其中86%是人类特有的,88%是小鼠特有的。ROIs的平均长度为1-1.5kb,这一结果超越了之前有注释的lncRNA的平均长度(约0.5kb)。
捕获表现主要体现在两个方面:一是中靶(on-target)率,即源于探针区的读取比例,二是富集程度,或者说是捕获后中靶率的增加。为此,研究人员用MiSeq分别测序了捕获前和捕获后的文库。结果显示CLS的中靶率(29.7%/16.5%)和富集程度(19倍/11倍)与此前基因间区段lncRNA的短读长研究相当。虽然与cDNA片段中靶率相当,但是,与基因组DNA捕获相比,CLS还有不足,意味着在捕获效率上长cDNA片段存在损失。
2003年作为编码“DNA元素百科全书”项目的一部分, Roderic
Guigó成立了迄今为止最有影响力的人类和老鼠基因组数据库之一“GENCODE”。本文,研究人员用CLS改善了GENCODE。在Guigó和合作者们共同努力之下,已经完成了基因目录的大量改进工作,尤其是lncRNAs部分。“在这项研究中,我们生成了人类和小鼠3500个新长非编码RNAs的详细图谱(金色),占目前已知的所有lncRNAs(TOTAL)的20%。我们还表征了lncRNAs的基因组特征以了解这些非编码基因的工作原理,”共同一作、CRG研究员Julien
Lagarde和Barbara Uszczynska说。
“人类基因组中98%DNA都不编码蛋白质,距离我们完全理解它们的功能和在疾病中的角色还有很长的路要走。为了达到这个目标,我们需要完整的基因组图谱。我们的新方法朝这一方向迈出了重要一步,”CRG校友、目前是伯尔尼大学的首席研究员、本文共同领导者Rory
Johnson说。
“这种更便宜、更快和更准确的目录优化方法将首先受益全世界的科学家,继而改变世界,”Guigó总结道。
原文标题
High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing












