
分子靶向抗肿瘤药物药动学/药效学结合模型研究进展
周小庭,刘昊晨,何华,柳晓泉
*
(中国药科大学药物代谢动力学研究中心,江苏 南京210009)
[
摘要]
分子靶向抗肿瘤药物可选择性抑制与肿瘤生长和增殖相关的酶、信号通路及生长因子受体,从而发挥抗肿瘤效应。因其具有疗效显著、对正常组织毒副作用小且耐受性好等优点,已成为癌症治疗领域的研究热点。药动学/药效学(PK/PD)结合模型作为定量描述“血药浓度-时间-效应”三者之间相互关系的有力工具,近年来被广泛应用于分子靶向药物研究,对于阐明药物作用物质基础、加速新药研发进程、优化给药方案、实现个体化给药等诸多方面具有重要意义。综述分子靶向抗肿瘤药物的PK/PD 模型研究进展及其应用现状与前景。
[
关键词]
分子靶向药物;PK/PD 模型;新药研发;优化给药方案;个体化给药
分子靶向药物选择性作用于肿瘤细胞,对正常组织损伤小且耐受性良好,已成为当前癌症治疗领域的研究热点。目前已有诸多分子靶向药物应用于临床,根据其来源和作用机制主要分为小分子激酶抑制剂、抗体靶向药物,以及抗体偶联药物(
ADC
)、以程序性死亡受体
1
(
programmeddeath 1
,
PD-1
)为靶点的肿瘤免疫疗法等近年来出现的新型分子靶向药物。分子靶向药物为恶性肿瘤的治疗带来新的希望,然而由于分子靶向药物体内药物浓度与药效之间关系极为复杂,如何评价在不同给药剂量下的疗效、毒性是分子靶向药物研究的一大难点。
药动学
/
药效学(
pharmacokinetics/pharmaco-dynamics
,
PK/PD
)结合模型作为定量反映“血药浓度
-
时间
-
效应”三者间关系的有力工具,可有效揭示分子靶向药物
PK
与
PD
之间的相互关系,目前广泛应用于药物的早期研发及临床给药方案的优化。本文对分子靶向抗肿瘤药物
PK/PD
结合模型的研究进展及其应用现状与未来前景作一综述。
1
常用于靶向抗肿瘤药物研究的PD 模型
根据应用范围的不同,常用分子靶向抗肿瘤药物研究的
PD
模型有
3
类:
1
)肿瘤生长抑制模型,用于描述肿瘤的生长状况;
2
)总存活数模型,用于描述肿瘤患者的最终获益;
3
)间接效应模型,用于表述和预测肿瘤及药物反应相关生物标志物的变化。
1.1
肿瘤生长抑制模型
近年来,描述和预测肿瘤生长过程的数学模型取得了突破性研究进展,主要包括
Simeoni
等
提出的预测性
PK/PD
模型(见图
1
)和
Claret
等
提出的肿瘤生长抑制模型(
the tumour growthinhibition model
,
TGI model
),前者主要用于预测临床前异种移植瘤小鼠的肿瘤生长及药物的抑制作用,而后者可同时应用于描述临床前和临床不同类型肿瘤的生长及对不同抗癌药物的反应。
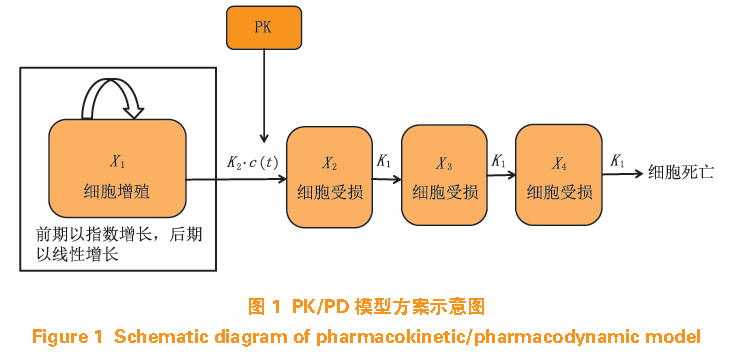
图1 的模型假设在无药物干预条件下,肿瘤细胞的生长期包括早期的指数生长期和后期的线性生长期,而在药物干预下,其中一部分细胞停止增殖,甚至死亡;xn(t) 表示某一时期部分增生的细胞,k1 为细胞凋亡一级速率常数,k2 表示药物效率常数,c(t) 表示血浆药物浓度,λ0、λ1 分别表示肿瘤在指数增长期和线性增长期的速率常数,Ψ 为调节系数,ω(t) 为肿瘤质量随时间变化的函数,ω0 表示刚接种时的肿瘤质量。该模型的微分方程见公式(1)~(7):
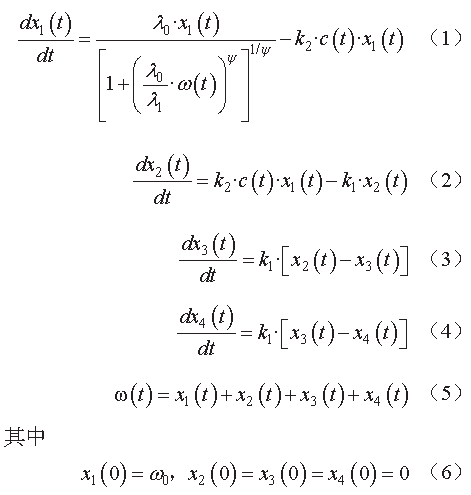
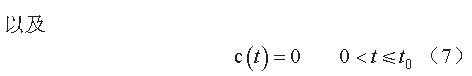
该模型已成功应用于贝伐单抗(
bevacizumab
)临床前给药方案设计,埃罗替尼(
erlotinib
,
ER
)临床前药物效应研究,埃罗替尼和舒尼替尼(
sunitinib
,
SU
)联合用药的疗效评价
以及
ADC
类药物临床前药效、毒效
等靶向抗肿瘤药物的研究。该模型具有较强的灵活性,不仅可反映自然状态下肿瘤在指数生长期、线性生长期的实际生长状况,还可用于预测不同给药方案下药物暴露量和肿瘤反应的关联情况;其局限性是只适用于临床前异种移植瘤小鼠,不适用于临床肿瘤患者。
Claret
等
在前人研究的基础上提出
TGI
模型,其表达式见公式(
8
)~(
9
):
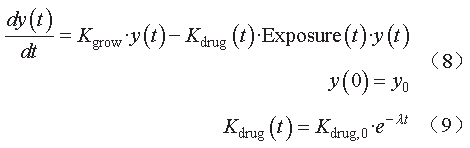
其中,
K
grow
为无药物干预条件下肿瘤自然生长的一级速率常数;
y
(
t
)
表示肿瘤的最大直径和(
sum of thelargest diameter
,
SLD
)随时间的变化;
K
drug
为药物对肿瘤的杀伤率常数;
Exposure
为药物的暴露水平,可以用给药剂量、血药浓度
C
或
AUC
表示;
e
-
λt
表示由耐药性导致的药物效应的衰减程度,参数
λ
也可反映不同肿瘤对药物的敏感程度。
TGI
模型将肿瘤的
SLD
作为肿瘤反应和药物的效应指标,不仅适用于临床前异种移植瘤小鼠模型,还可进一步推广用于临床研究;同时,该模型还引入反映肿瘤对药物敏感程度参数
λ
,考虑了肿瘤耐药性的影响,故更符合实际情况。然而,在很多情况下血浆中的药物暴露水平并不能很好地反映药物抗肿瘤效应,循环系统中生物标志物的变化往往有更好的预测效果。因此,在靶向药物研究中,
Hansson
等
将基于机制的生物标志物参数引入
SU
的
TGI
模型,如公式(
10
)~(
12
)所示:
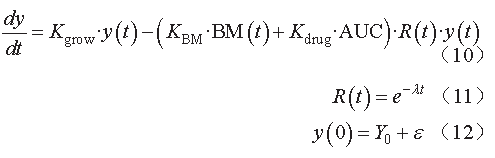
其中,
K
BM
代表与生物标志物(
sKIT
、
sVEGFR-3
)变化相关的肿瘤缩减速率常数;
ε
为残差。
1.2
总存活数模型
总存活数(
overall survival
,
OS
)是临床上评价抗肿瘤药物效益的首选终指标,
OS
模型是基于临床观察数据建立的描述存活时间(
T
)分布的函数(包括对数正态分布、
Weibull
分布、
Logistic
分布、
Log-logistic
分布和指数分布等),用于预测某一时间出现死亡事件的概率。风险函数
h
(
t
)
表达式如公式(
13
)所示。
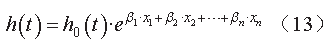
h
0
(
t
)
为基线风险,可通过一个或多个估算参数来定义;
x
1
、
x
2
……
x
n
代表一系列预测因子(可以是与肿瘤大小、肿瘤生长、生物标志物等相关的参数);
β
1
、
β
2
……
β
n
分别为相应预测因子的回归系数。在针对舒尼替尼的研究中,
Hansson
等
分别将生物标志物(
VEGF
、
sVEGFR-2
、
sVEGFR-3
、
sKIT
)和不良反应(骨髓抑制、高血压、疲劳、手足综合征)等作为预测因子分别代入
OS
模型当中,从而选择出可靠的预测因子,最终得出
OS
模型如公式(
14
)、(
15
)所示。
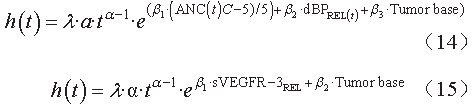
公式(
14
)、(
15
)均以
Weibull
分布描述基线风险,其中
λ
为风险系数,
α
为形状因子,
ANC(
t
)
为中性粒细胞随时间减少的函数。
将生物标志物、不良反应等纳入到
OS
模型中,能有效预测肿瘤存活情况,并可根据Ⅱ期临床试验数据预测Ⅲ期试验结果。但由于
OS
模型是固定时间点的函数,因此,不适用于药物在评估时间点之后对肿瘤改变情况的预测。
1.3
间接效应模型
从机制上看,分子靶向药物是通过作用于特定的致癌位点,继而引起内源性物质含量或活性的改变,从而间接地发挥抗肿瘤效应。此内源性物质即所谓的生物标志物,生物标志物在靶向药物研究中可作为替代终指标,对药物疗效或毒性效应进行早期预测,同时也可用于对抗癌药物作用机制的进一步探讨。间接效应模型(
the indirect response model
,
IDR model
)能很好地表征靶向药物机制相关生物标志物与药效之间的关系,是目前靶向药物研究中应用较为广泛的一类数学模型。
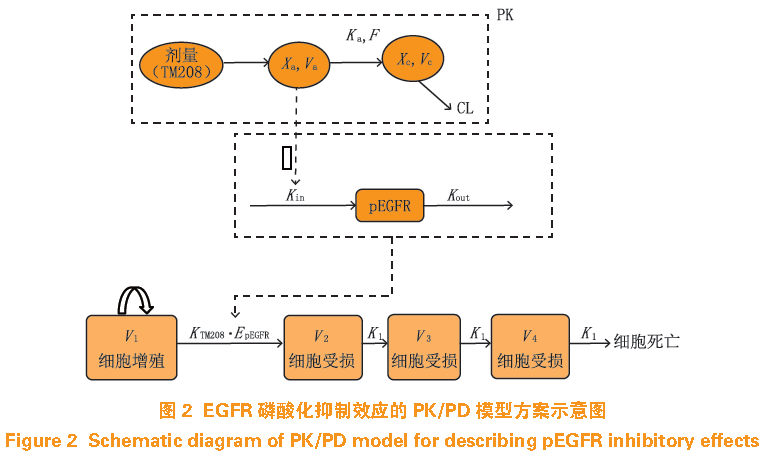
TM208
是一种新型的
EGFR
酪氨酸激酶抑制剂(
EGFR-TKI
),通过抑制
EGFR
的磷酸化抑制作用(
pEGFR
)发挥抗癌疗效。
Ji
等
以
IDR
模型表述
TM208
血药浓度与
pEGFR
之间的关系(见图
2
),模型方程见公式(
16
)。
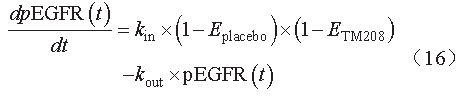
由于
IDR
模型是基于药物作用机制而建立的
PK/PD
模型,因而可用于充分了解机体与药物疗效及不良反应之间的相互关系。然而,此模型的局限性在于需对肿瘤及药物的作用机制充分了解,并提供一个或多个在实验过程中可连续测定的替代终指标,但在临床中获得替代终指标数据较为困难。
PK/PD
模型的应用已贯穿于药物研究的各个阶段,上述
3
种模型在不同研究阶段发挥各自作用,在实际应用中可根据药物的作用特点加以选择使用。但需要指出的是,上述
3
种模型虽是应用于肿瘤研究的不同方面,三者往往联合使用,
IDR
模型参数可用于表述肿瘤生长抑制或存活数情况(见图
2
),从而通过构建的整体
PK/PD
模型更好地表述药效关系。此整体建模方法已广泛应用于
SU
的临床研究
及
ER
、
PF06463922
、
PF06471404
和克唑替尼(
crizotinib
)等新型分子靶向药物的临床前机制探讨和药效研究。
2 PK/PD
模型在靶向抗肿瘤药物早期研发中的应用
PK/PD
建模作为传统生物医学研究模式的补充,是药物研究中重要的辅助工具,在药物早期研发阶段,可用于临床前给药剂量的选择与最优化试验设计、预测临床有效剂量、发现潜在的药物作用靶标、预测靶组织中药物浓度等诸多方面。加速新型化合物在体内活性的评价,并可将临床前研究工作外推到临床,达到可靠预测的目的,减少肿瘤临床试验的失败率,且可有效降低药物研发成本。
2.1
用于预测药物在肿瘤组织中摄取和分解代谢过程
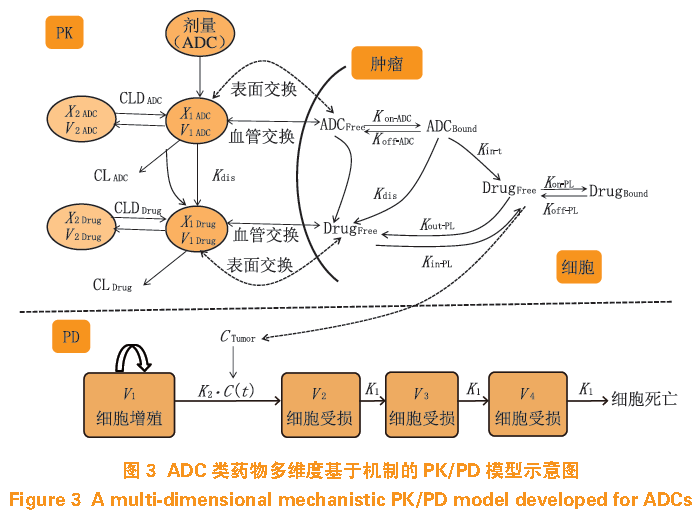
肿瘤生理结构(如细胞间隙大小、血管结构等)不同于机体正常组织或器官,大多数情况下肿瘤组织中药物浓度与血浆中药物浓度是不平衡的,一般通过经验来推测两者之间的相互关系,但这种基于经验的方法往往并不可靠,因此建立可靠的数学模型表述靶组织中药物浓度与抗癌效应的关系尤为必要。以
ADC
类药物为例,此类药物通过化学键将细胞毒剂与单克隆抗体连接,从而靶向递送化学治疗剂直接作用于癌组织,与传统的化疗药物相比其作用机制更加复杂。
Singh
等
建立了用于同时表述
ADC
类药物在血浆和肿瘤中处置过程的肿瘤处置模型,并与
Simeoni
的
TDI
模型相结合创建
PK/PD
模型,用于研究肿瘤中药物浓度与抗癌效应之间的关系(见图
3
)。
2.2
用于早期研发阶段分子靶向药物活性筛选
PF06463922
和
PF06471402
是新型间变性淋巴瘤激酶(
anaplastic lymphoma kinase
,
ALK
)抑制剂,应用于对第
1
代
ALK
抑制剂
crizotinib
抵抗的非小细胞肺癌(
NSCLC
)的治疗。为了描述和比较
PF06463922
与
PF06471402
在移植瘤小鼠模型中对靶标的调节作用及抗肿瘤活性的区别,
Yamazaki
等
用数学建模的方法展示了在小鼠模型中两者对
ALK
的调节作用及
PD
差异,给药方式分为经口给药和皮下注射,
PK
模型均以一房室模型拟合,并以
IDR
模型表述血药浓度与
ALK
磷酸化抑制作用(
pALK
)之间的关系,计算结果显示经口给药条件下两者的
EC
50
分别为
36
和
20 nmol
· L
-1
、皮下注射给药的
EC
50
分别为
100
和
69 nmol
· L
-1
。药物
-
疾病模型显示,经口给药时使肿瘤生长停滞的药物浓度分别为
51
和
27 nmol
· L
-1
;皮下注射给药时肿瘤生长停滞的药物浓度分别为
116
和
70 nmol
· L
-1
。表明,在不同给药方式下,药物对靶标的调节作用与药物的抗肿瘤效应之间均为高度相关,因此可以利用对
ALK
的抑制作用来评价药物的抗肿瘤效应,且
PF06471402
的抗肿瘤效应强于
PF06463922
。
2.3
用于早期研发阶段分子靶向药物决策过程
PK/PD
模型应用于靶向药物研发的早期决策过程,在临床前实验的最优化设计中,可用于
PD
、急性毒性、重复给药毒性的评估,预测药物进入人体研究的初始剂量和给药方案。在Ⅰ期临床研究阶段,不仅可进行人体
PK
及药物耐受性的初步评估,以确定不同给药方案下剂量限制性毒性(
DLT
)和最大耐受剂量(
MTD
),还可进行药物对靶点抑制效果的探讨,从而确定下一步研究计划。
Baselga
等
在新型酪氨酸激酶抑制剂
AEE788
的Ⅰ期临床研究中,以
Emax
模型表征药物暴露量与相关通路生物标志物之间的关系,以剂量递增方法得出
MTD
为
450 mg
· d
-1
,在
MTD
剂量范围内对
pEGFR
有抑制作用,但对
pVEGFR
抑制作用不大,不能达到很好的抗肿瘤效应,且易耐受、具有肝毒性等缺点,因此终止对
AEE788
进一步临床研究。
Gueorguieva
等
利用
PK/PD
模型探索新型转化生长因子
β
(
TGF-
β
)抑制剂
LY2157299
的临床治疗窗,其中以
TGF-
β
激酶磷酸化百分比(
pSMAD
)为药效指标,以
Emax
模型表征药物暴露量与
pSMAD
之间的关系,根据临床前
PK/PD
模型模拟不同给药剂量和给药时间下
pSMAD
与毒性的变化,推断出其临床安全有效给药剂量为
160
~
360 mg
· d
-1
。
3 PK/PD
模型在分子靶向抗肿瘤药物最优化给药方案研究中的应用
靶向药物的出现在一定程度上提高了恶性肿瘤的治疗效率,然而在治疗剂量下出现的药物毒性和耐受性等问题仍然是限制其在临床上广泛使用的主要原因。
PK/PD
模型在临床最优化给药方案研究中,可用于最佳给药剂量、给药时间的选择,使药物治疗达到最大临床受益的同时降低毒性和耐受性的发生。
3.1
用于临床最佳剂量估算
PK/PD
模型可以利用少量可获得的临床试验数据进行建模和仿真,模拟不同给药剂量下的药效和毒效反应,从而实现最佳给药剂量估算。
Houk
等
收集
639
例服用
SU
的胃肠道间质瘤(
GIST
)、转移性肾细胞癌(
mRCC
)患者数据以建立群体
PK/PD
模型,其中以肿瘤进程时间(
TTP
)和总存活数(
OS
)为药效指标,以疲劳发生率、中性粒细胞绝对计数(
absoluteneutrophil count
,
ANC
)、舒张期高血压为毒效指标,并在上述模型的基础上模拟不同给药剂量下的药效、毒效反应,结果显示
50 mg
· d
-1
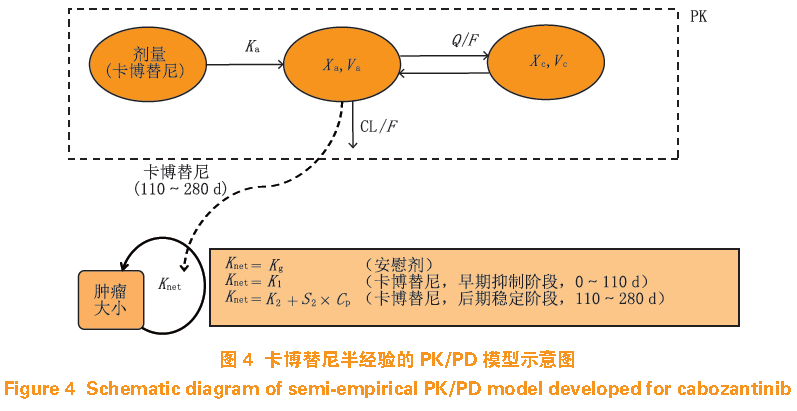
的初始剂量可使患者临床受益,并将不良反应发生率控制在可接受范围内。另一项研究通过收集甲状腺髓样癌患者服用卡博替尼(
cabozantinib
)的Ⅲ期临床试验数据,以半经验的
PK/PD
模型拟合卡博替尼药物暴露量与肿瘤生长之间的关系(见图
4
)。该模型包括服药早期的癌细胞数量减少期(
0
~
110 d
)和服药后期的稳定期(
110
~
280 d
),很好地表述了卡博替尼具有杀死癌细胞以及抑制癌细胞生长的双重作用。对卡博替尼的给药剂量与抗癌效应的模拟结果显示,在服药早期、服药后期由原来的
140
、
100 mg
· d
-1
的推荐剂量改为
100
、
60 mg
· d
-1
,能达到临床给药条件相当的抗癌效果。
3.2
用于确定临床最佳给药时间
通常抗肿瘤药物的疗效和安全性与给药方案密切相关,不同的给药方案(如给药间隔和给药剂量等)可能产生不同的毒性反应。以
SU
为例,在其临床推荐剂量(50 m
g
· d
-1
)连续服药4 周停药2 周(4/2 给药方案)下,仍具有显著毒性,从而不得不降低给药剂量或中断给药。然而,维持一定的药物浓度是实现最佳药物疗效的必然前提,降低给药剂量往往不能达到有效的治疗效果,因此给药时间和给药间隔也是临床
研究中需要考虑的因素。为了比较
SU
连续服药
2
周停药
1
周(
2/1
给药方式)与传统给药方案的差异,
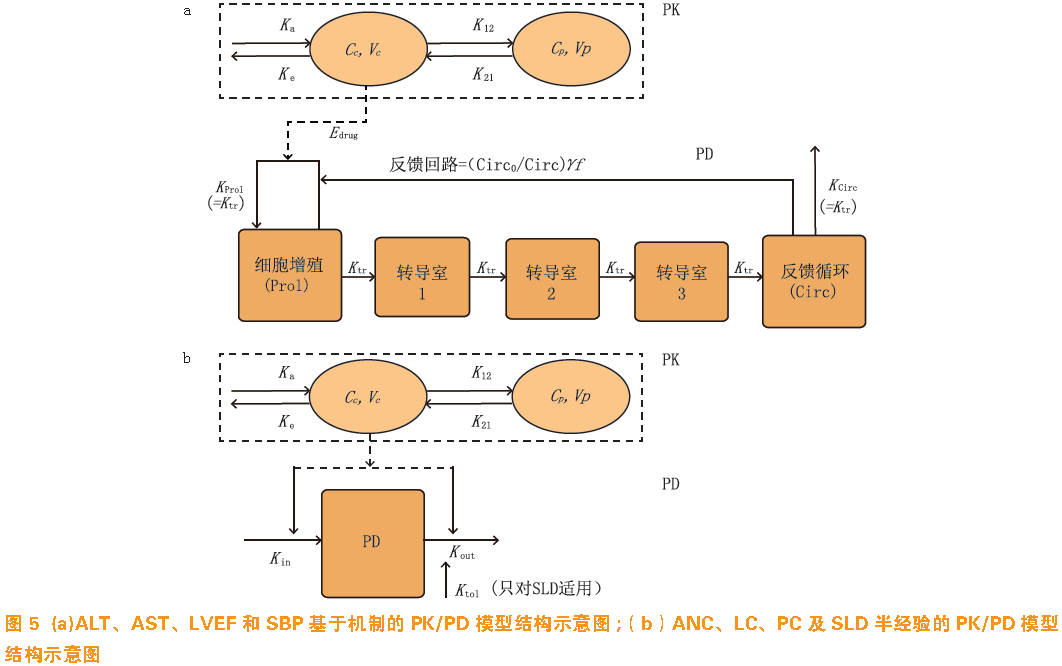
Khosravan
等
以肿瘤
SLD
为药效指标,以
ANC
、血小板计数(
plateletcount
,
PC
)、淋巴细胞计数(
lymphocyte count
,
LC
)、丙氨酸转氨酶(
alanineaminotransferase
,
ALT
)、天冬氨酸转氨酶(
aspartateaminotransferase
,
AST
)、舒张压(
diastolicbloodpressure
,
DBP
)、左心室射血分数(
left ventricularejection fraction
,
LVEF
)为毒效指标,建立基于机制的
PK/PD
模型(见图
5a
)以及半经验
PK/PD
模型(见图
5b
)用于预测给药方案下药物安全性、有效性的差异,结果显示
2
种给药方案的抗癌效应相当,但2/1给药方案所引起的血小板减少的毒副作用较小,提示2/1 给药方式优于目前临床推荐的4/2 给药方式,具有更好的耐受性。
3.3
用于给药方案个体化及调整
随着基因组学、转录组学、蛋白质组学和代谢组学等分子分析技术的发展,人们逐渐认识到癌症具有个体化差异,因此催生出个体化治疗新策略,即根据基因组分型的不同选择具有针对性、有效的化学疗法。个体化治疗策略的实施依赖于以基因差异为基础、以蛋白质为中心的药物作用动态网络模型的建立,
Iyengar
等在传统
PK/PD
模型的基础上,将系统生物学融入到
PD
建模中从而提出增强型
PD
模型(
enhanced PD model
,
ePD
)。
ePD
模型在药物调节网络分析中的应用可以解释多靶点以及基因组、表观遗传、翻译后修饰等差异对药物效应的影响,推动药物研发,实现精准医疗。
在癌症治疗中,血管内皮生长因子受体(
vascularendothelial growthfactor receptor
,
VEGFR
)信号通路与肿瘤血管生成密切相关,
VEGF
与
VEGFR
结合激活下游的细胞外信号调节激酶(
ERK
)和丝氨酸/苏氨酸蛋白激酶(
Akt
),从而导致血管内皮细胞等扩散和生存,复杂的
VEGFR
调节网络也为靶向治疗提供多个药物介入靶点。
Zhang
等
根据现有的研究建立
VEGFR
基础网络模型,并利用建模与仿真初步筛选出能表征生物学意义的动力学、蛋白质丰度等参数,然后将
SU
的
PK
模型与此网络模型建立连接,创建出
PK/ePD
模型;通过全局灵敏度分析显示,对
SU
药效贡献最大的为
VEGFR
,磷脂酰肌醇
3-
激酶(
PI3K
)次之, 丝裂原活化蛋白激酶激酶(
MEK
)、
MEK
激酶(
Raf
)、磷脂酶
C
γ
(
PLC
γ
)也有重要作用,可确定为潜在的药物作用靶标;在随后的研究中选择阻断
VEGF
与
VEGFR
结合的
bevacizumab
、
PI3K
抑制剂
BKM120
、
MEK
抑制剂
AZD6244
和
PLC
γ
抑制剂分别进行最优化疗策略的研究;以连续
28 d
实现
80%
的
pERK
和
pAkt
抑制为目标,模拟了
SU
不同给药条件下联合使用上述
4
种药物给药的剂量和时间,从而提出最佳给药方案。
3.4
用于不良反应的评估及预测
分子靶向化疗药物通过抑制与疾病相关的一个或多个靶点而发挥抗肿瘤效应,然而当特定靶点被抑制的同时,也会导致与该靶点相关的生理通路的紊乱。例如作用于
VEGF
信号通路的靶向药物在抑制肿瘤血管生成的同时,阻断
VEGFR
也将致使下游调节血压相关的内皮素系统以及
NO
系统的紊乱,引起血压(
BP
)升高、蛋白尿(
proteinuria
,
PU
)等不良反应。
PK/PD
模型在这些药物的临床和临床前不良反应的
评估和预测中均发挥重要作用,
Keizer
等
收集了
67
例受试者中服用
E7080
(一种新型的抗血管生成药物)的药物暴露量、
BP
和
PU
等Ⅰ期临床试验数据,分别以
IDR
模型和
Markov
转换模型拟合了
BP
、
PU
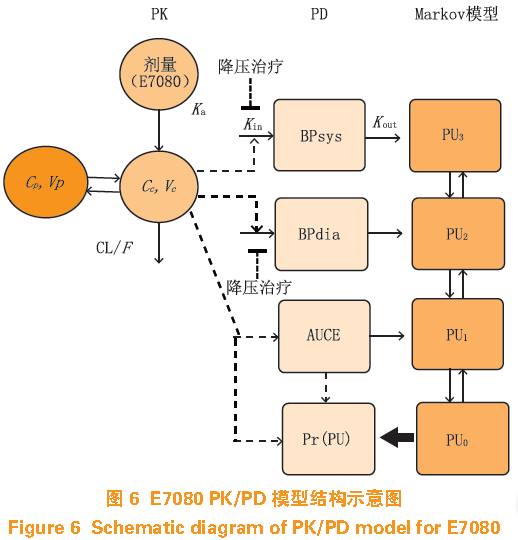
等不良反应与药物之间的量效关系(见图
6
)。该模型可进一步用于其他
VEGF
通路抑制剂引起的
BP
及
PU
的评估与预测。
3.5
用于靶向药物联合用药评价
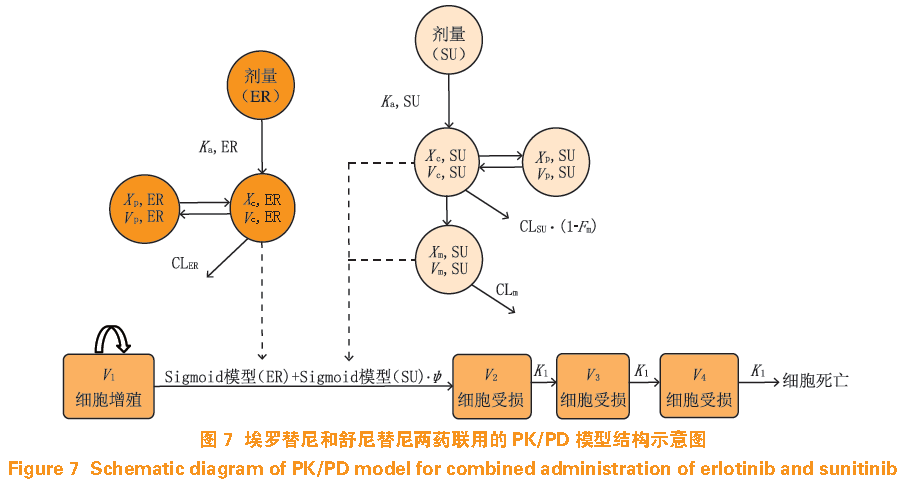
联合用药在肿瘤治疗中具有诸多优势:一方面,不同作用机制的靶向药物联用可抑制肿瘤多条信号通路,达到更好的治疗效果;另一方面,可减少机体对药物的耐药性;此外,联合治疗中单一组分的给药剂量减少,相应的毒性效应也降低。PK/PD 模型在联合用药中可用于定量描述不同给药条件下多药联用的药效、毒效改变,判断是否存在协同作用,并对联用方案进行优化。Li 等
在人体A549 NSCLC 异种移植瘤小鼠中研究了EGFR 抑制剂ER 与VEGFR 抑制剂SU
2
个靶向药物不同剂量下单独给药与联合用药的疗效,每天记录肿瘤大小数据,并以二房室PK 模型合并Simeoni TGI 模型定量描述血药浓度与肿瘤抑制之间的关系,两药联用的PK/PD 模型结构如图7 所示。结果表明,联合用药组疗效远远强于单一用药组,提示ER、SU 具有强协同作用;通过模拟不同给药剂量下的联合效益,提示在联合治疗中SU 浓度起决定作用,ER 低剂量(≤5 mg
·
kg
-1
)条件下,调节SU 的剂量可达到最佳治疗效果。
4
结语
随着科学技术的发展,越来越多的分子靶向药物被批准用于恶性肿瘤的临床治疗,尚有更多的新型分子靶向药物处于开发阶段。近年来,
PK/PD
建模与仿真在靶向药物研究中已得到广泛的应用,加快了药物的研发进程,并促进临床合理用药研究。然而,在肿瘤
PK
的表述以及血药浓度与药效、毒效相互关系的认识等方面尚不完全清楚,限制了
PK/PD
模型的使用。为了克服上述困难,仍需要更多的实验和建模尝试,从而构建出基于肿瘤特殊结构以及作用机制的模型,扩大
PK/PD
模型在分子靶向药物研究中的应用。

[
专家介绍]
柳晓泉:
教授,中国药科大学药物代谢研究中心博士生导师,现任国家和江苏省药品审评专家,江苏省青蓝工程培养对象。长期致力于药物代谢动力学基础理论和应用的研究,以药物代谢动力学新理论、新模型、新技术和新方法的研究及其在新药开发研究中的应用为主要的研究方向,主持
3
项国家自然科学基金。
2004
年获得江苏省科技进步一等奖
1
项(创新药物体内吸收、代谢、分布与排泄新理论与新模型研究);
2007
年获得国家科技进步二等奖
1
项(临床前药物代谢动力学关键技术与研究体系)。在国内外学术刊物上发表有关的学术论文
100
余篇。
●
感谢您阅读《药学进展》微信平台原创好文,也欢迎各位读者转载、引用。本文选自《药学进展》2017年第8期。
●
《药学进展》是一本专注于医药领域前沿动态的专业媒体,月刊,铜版纸全彩印刷,全年360元,欢迎订阅!编辑部官网:www.cpupps.cn;电话:025-83271227。




