前期文章
一个超简单的转录组项目全过程--iMac+RNA-Seq(一)
一个超简单的转录组项目全过程--iMac+RNA-Seq(二)QC
3 Alignment
有很多软件都可以比对到参考基因组,hisat2,bowtie2,STAR等等(手上这台iMac不够内存跑STAR)
首先学一下hisat2的用法
hisat2 --help ## 主要要学会使用--help等参数调用帮助文档(软件说明书)
Usage:
hisat2 [options]* -x <ht2-idx> {-1 -2 |
-U | --sra-acc <SRA accession number>} [-S ]
-p/--threads number of alignment threads to launch (1)
用法一,一步一个脚印
## 用“指鹿为马“的方法,将会重复出现的路径等缩略
wkd=/Users/Desktop/project
## 比对
cd $wkd/clean
ls *gz|cut -d"_" -f 1,2 |sort -u |\
while read id;
do
ls -lh ${id}R1_val_1.fq.gz ${id}R2_val_2.fq.gz
hisat2 -p 4 -x \
/Users/Desktop/project/reference/hg38/hisat2 \
-1 ${id}1_val_1.fq.gz \
-2 $
{id}2_val_2.fq.gz -S ${id}.hisat.sam
done
## 转为二进制文件sam to bam
ls *.bam| while read id;
do
(samtools sort -@ 4 -o $(basename ${id}".sam").bam ${id});
done
rm *.sam # 移除sam文件
用法二:一步到位
vim align.sh ## 创建一个脚本,写入以下内容
#!/bin/bash
set -e
set -u
set -
o pipefail
# source activate rna
# set PATH
HISAT2=/Users/Desktop/project/reference/hg38/hisat2
pwd
ls *gz | cut -d"_" -f 1,2 | sort -u |\
while read id
do
echo "processing ${id}_R1_val_1.fq.gz ${id}_R2_val_2.fq.gz"
hisat2 -p 4 -x \
$HISAT2/hg38.ht2 \
-1 ${id}_R1_val_1.fq.gz \
-2 ${id}_R2_val_2.
fq.gz |\
samtools sort -@ 4 > ${id}.hisat.bam
done
# source deactivate
运行脚本
nohup bash align.sh &
查看结果
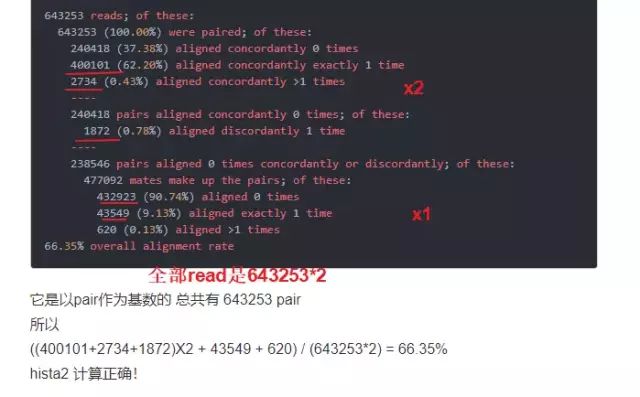
请关注比对报告中的aligned concordantly exactly 1 time这一项内容,至少要80 %以上。
小彩蛋
:洲更老师的锦囊 jobs and disown的使用
当我忘记nohup脚本的时候,但我又要离开实验室了,这时候使用disown帮助把跑到一半的任务丢到后台,惊呆了!神仙走位!
小结:本章所需知识背景
(1)什么是比对,比对前后数据的变化;
(2)fasta,fastq文件的规则以及包含的信息;
(3)比对结果的查看,比对率的由来。
以上知识点均可以在网上查到,建议在进行着一部分项目的时候,一定要补充上最基本的知识点。
问题集锦
收到了几个小问题,不过我几乎都无法解答,但我可以找到答案~
我的怎么QC怎么回事?跑着跑着跑着就停了呀。
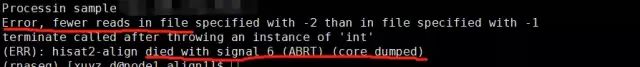
QC报错?
问了代码才知道问题可能出在input上,过滤结束后的样本名是val.fq.gz
,出现这样结果的原因,应该是过滤出错了。
for id in {xx1,xx2,xxx3};
do
echo "Processin sample ${id}"
hisat2 -p 5 -x /data1/reference/index/hisat2/hg19/hg19 \
-1 /data1/projects/clean/${id}_R1_trimmed.fq.gz \
-2 /data1projects/clean/${id}_R2_trimmed.fq.gz \
-S /data1/projects/align1/${id}.hisat.sam
done
为什么比对率这么低?
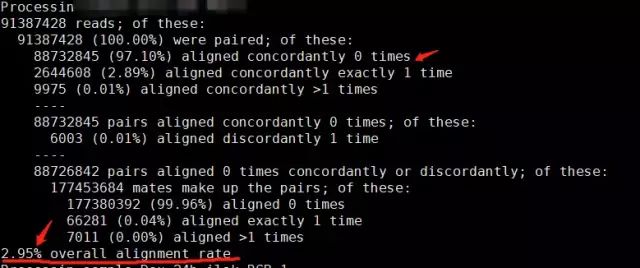
解答:

比对率的计算好像不对?
怎么加起来都不等于96.7%呀?这样的问题,一般我都解决不了,我会直接放弃,然后寻求帮助。

解答:
具体请看简书:2018-12-04 Hisat2 map 结果与 samtools flagstat 结果不一致