
最近,辉瑞新一代ALK/ROS1酪氨酸激酶抑制剂Lorlatinib被FDA授予突破性药物资格。于是,Lorlatinib作为第三代ALK抑制剂进入大家的视野,也为病人带来了新的希望。
当看到Lorlatinib的结构(图1),其大环结构对我来说成了一个谜,为什么是一个大环结构呢?这种结构又是怎么被发现的?Lorlatinib又为什么会被定义为第三代ALK抑制剂?带着这些疑问,我试图为自己以及与我有着同样疑惑的朋友解读Lorlatinib的大环之谜。
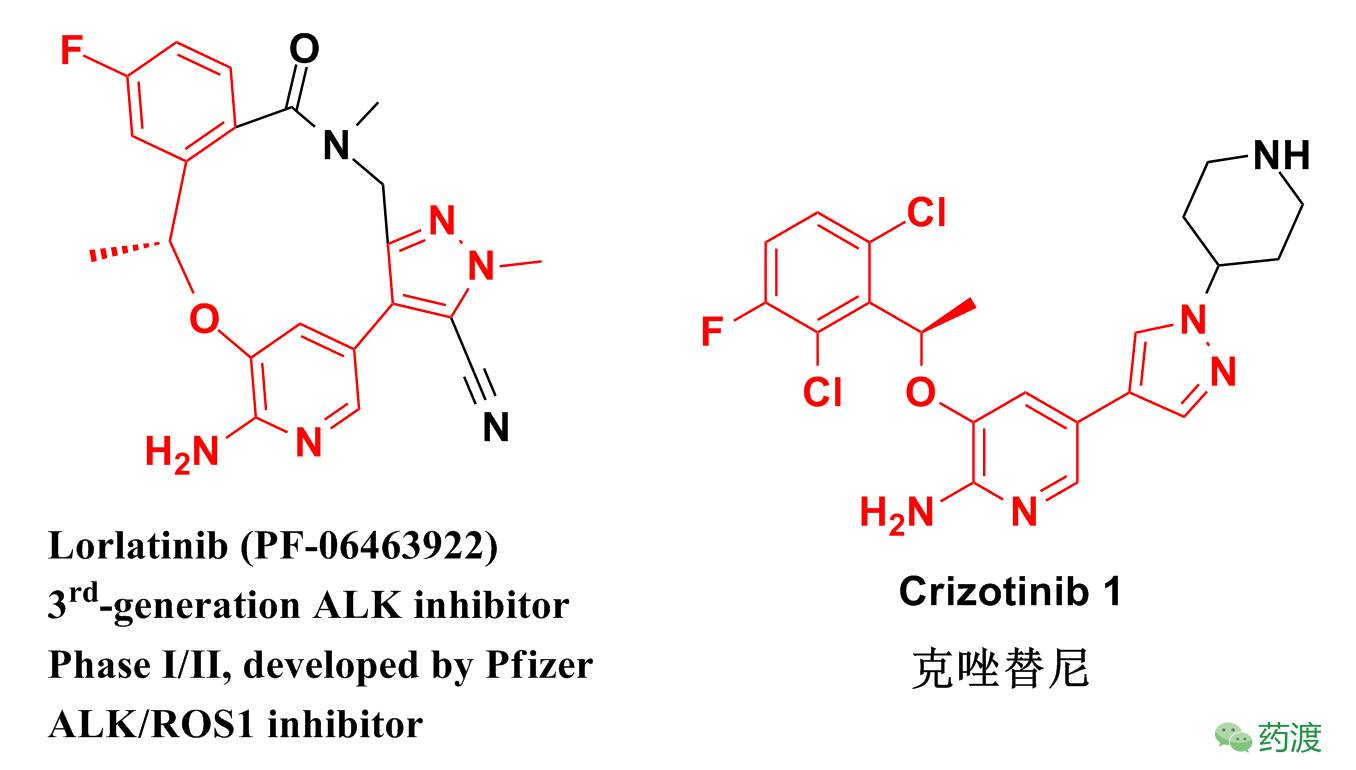
图 1
填补尚未满足的医疗需求是新药研发的一个重要动力,也是衡量一个新药上市后在商业上能否取得成功的重要评估指标。Lorlatinib正是为未满足的医疗需求而生。Lorlatinib之所以被称为第三代ALK抑制剂,还要从ALK抑制剂的发展说起。
间变性淋巴瘤激酶(Anaplastic Lymphoma Kinase,ALK)是一种受体酪氨酸激酶,与血液、间质和实体三大类型肿瘤相关。约3-7%非小细胞肺癌(NSCLC)患者体内肿瘤染色体EML4基因外显子与ALK基因外显子融合,形成EML4-ALK融合酪氨酸激酶,EML4-ALK融合变异体具有高度的致癌性,且ALK在多种肿瘤细胞中高表达。因此,ALK成为一个极具吸引力的癌症治疗靶点。
辉瑞研发的第一代ALK抑制剂克唑替尼即是基于EML4-ALK靶点的发现应运而生。克唑替尼(Crizotinib)是一种ATP竞争性的多靶点蛋白激酶抑制剂,它可有效抑制MET/ALK/ROS的细胞生物活性,并分别在ALK、ROS或MET激酶活性异常的肿瘤患者中显示出较高的临床疗效。克唑替尼是肿瘤药物研发史上最快速的药物之一,2011年在美国上市后引起轰动。美国FDA在2016年又批准克唑替尼(Crizotinib)可用于治疗携带ROS-1基因突变的晚期(转移性)非小细胞肺癌(NSCLC)患者。
克唑替尼彻底改变了携带ALK染色体重组的晚期非小细胞肺癌(NSCLC)患者的治疗。不幸的是,大部分患者在治疗后的12个月内对克唑替尼产生耐药性,并发生获得性耐药突变,在发生中枢神经系统转移的患者中的疗效更是十分有限。这种情况催生了对克唑替尼耐药的NSCLC的第二代ALK抑制剂的快速发展。最近几年,已经不断涌现出多个疗效好、毒性低的第二代ALK抑制剂(图 2):Ceritinib、Alectinib、Brigatinib。然而,抗肿瘤领域激酶抑制剂的坎坷命运总是惊人的相似,那就是耐药性的产生使其失去有效性。最初,几个第二代ALK抑制剂对发生中枢神经系统转移的克唑替尼耐药的患者显示了治疗活性,不过顽固的肿瘤很快就对这些新抑制剂产生了耐药性。
面对克唑替尼及第二代ALK抑制剂的耐药,以及耐药后的无药可用,辉瑞的科学家们开始研发第三代ALK抑制剂,即可用于对克唑替尼及第二代ALK抑制剂耐药的、发生中枢神经系统转移的NSCLC患者仍有疗效的药物。辉瑞研发出的第三代ALK抑制剂即是Lorlatinib。
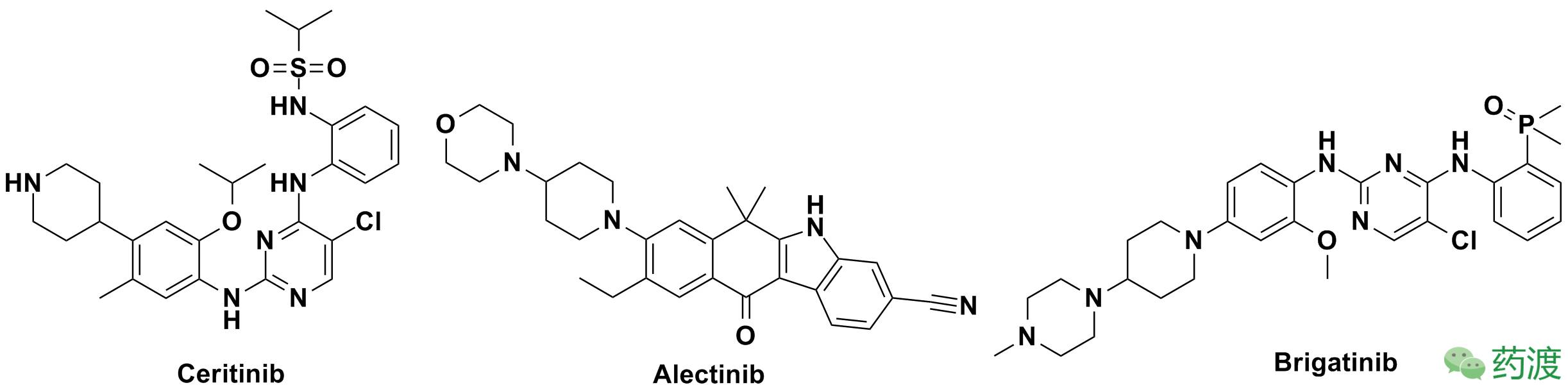
图 2. 第二代ALK抑制剂
当我们比对克唑替尼和Lorlatinib的化学结构时就会惊讶的发现,这两个隔代相望的ALK抑制剂结构上竟然有着非常大的相似性(图1)!为什么结构与克唑替尼具有很大相似性的Lorlatinib会成为第三代ALK抑制剂呢?这还要从Lorlatinib发现说起。
若想达到克服克唑替尼的耐药、提升药效、增加肿瘤细胞内和脑内游离药物浓度从而获得更好靶向治疗作用的目的,可以通过避免血脑屏障(BBB)和肿瘤细胞表面转运体介导的药物外排来增加中枢神经系统(CNS)内的药物含量。那么进一步提升克唑替尼的类药性质就成为化合物改造的重要目标。应用基于结构的药物设计策略和以亲脂性效率(LipE,是优化过程中监测化合物的活性、物化性质和成药性程度的一个指标,LipE=pKi (or pIC50) − logD)为重要优化指标,辉瑞科学家开始对克唑替尼进行结构改造,引入各种取代基团得到了一系列脂肪族类氨基吡啶和氨基吡嗪类ALK抑制剂。通过对A环和B环各种取代基团的引入(图3),发现了一个第二代ALK抑制剂类化合物PF-06439015。与克唑替尼相比,PF-06439015的活性得到大幅度提升,对野生型pALK 细胞抑制活性是克唑替尼的105倍,对突变型pALK-L1196M细胞的抑制活性是克唑替尼的128倍,并显示出较强的体内活性,PF-06439015的LipE (5.3)也比较适宜,但遗憾的是PF-06439015容易被外排(BA/AB = 30.6/2.8,ratio 10.9),脑内的游离药物浓度就会比较低,无法用于脑转移患者的治疗,因此,PF-06439015未用于进一步开发。
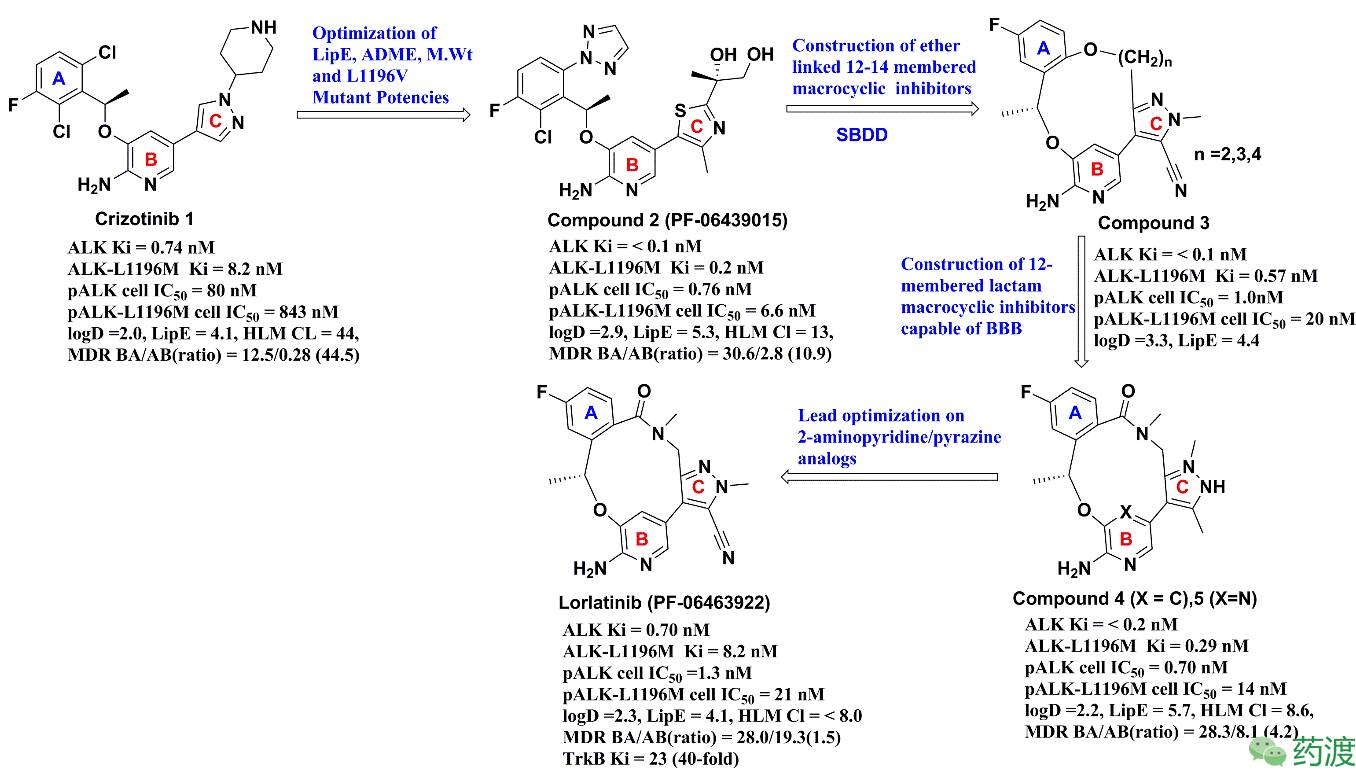
图 3. 从克唑替尼到Lorlatinibd的发现:
SBDD = Structure based drug design,BBB = Blood-brain barrier,LipE = lipophilic efficiency,ADME =Absorption,distribution,metabolism and excretion.
研究的脂肪类化合物不具备同时达到高活性(L1196M IC
50
<25nM)和低药物外排率(BA/AB < 2.5)的特性,难以提升药物在中枢神经系统(CNS)的有效性。因此,应用全新策略设计中性抑制剂成为一个新的研究方向。脂肪类化合物的研究提供了配体与蛋白相互作用的重要结构信息,如化合物A与ALK激酶的共晶结构所示(图4),化合物A的构象显示出分子首尾靠近的U形结构,结合其它配体与激酶相互作用的重要信息,研究者开始研究大环类结构抑制剂。大环类化合物一定程度上可以保留分子量大小、保留较好LipE,并可以实现很好的构象限制。
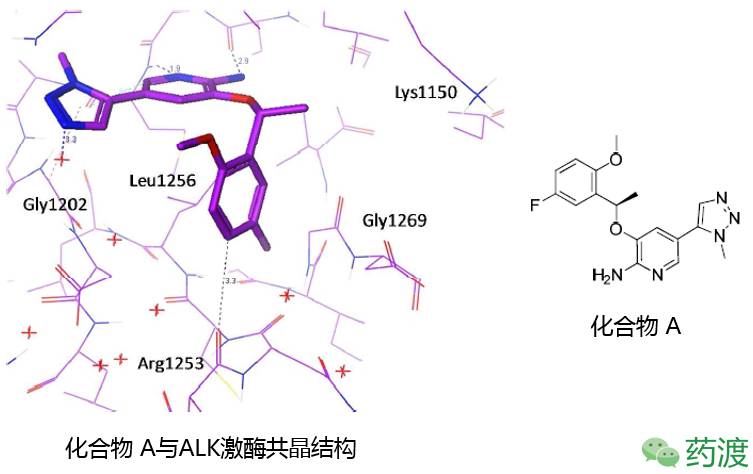
图4
研究者首先研究了含有醚链的12-14元环的大环结构化合物
3
(图3),但与脂肪族类化合物相比,化合物
3
脂溶性太高,活性也稍差,ADME和进入CNS的性能也存在问题。配体与蛋白的相互作用进一步指导设计在邻近吡唑位置引入酰胺得到12元的酰胺类大环结构化合物
4
(图3),降低了脂溶性、提升了LipE。与克唑替尼相比,酰胺类大环结构的活性与成药性都更加优异。因此,以酰胺类大环结构4为基础,进一步降低化合物被外排的性质(即降低BA/AB),提升脑内游离药物浓度成为化合物结构优化的重要方向。经过一系列结构优化,最终得到了含有腈基吡唑结构的酰胺类大环化合物PF-06463922,即第三代ALK抑制剂Lorlatinib(图3)。
Lorlatinib的发现过程远比在此呈现出来的复杂的多,它的发现也给我们进行新药研发给予了一些提示:
①新药研发的第一步是新药研发方向的选择,而好的新药研发方向是由尚未满足的医疗需求所主导的。
②启动新药研发项目首先必须确定明确的研发目标。正如研究第三代ALK抑制剂,辉瑞研究者所确定的研究目标就是发现能够作用于CNS的高活性ALK抑制剂,可用于对克唑替尼及第二代ALK抑制剂耐药的、发生中枢神经系统转移的NSCLC患者仍有疗效的药物。
③为研发目标的化合物确定关键参数。正如研究第三代ALK抑制剂,LipE和分子量(MW)被确定为需要优化的关键参数。配体与大多数蛋白的相互作用都受到亲脂性的影响。亲脂效率(LipE = pKi(或pIC50) -log D)作为每单位亲脂性的结合效力的数值指标,可被用于评估化合物优化的过程。旨在改善LipE的药物设计能够通过增加效能与亲脂性的比例,实现ADME的优化和增加药物安全性。而分子量多与渗透性呈现负相关,因此减小分子量对发现能够进入CNS发挥作用的成药性小分子至关重要。LipE和分子量(MW)这两个参数的确定对第三代ALK抑制剂Lorlatinib的发现过程中化合物的优化起到重要的指导作用。
参考文献
1.Johnson T W,Richardson P F,Bailey S,et al. Discovery of (10 R)-7-Amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4- (metheno) pyrazolo [4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922),a Macrocyclic Inhibitor of Anaplastic Lymphoma Kinase (ALK) andc-ros Oncogene 1 (ROS1) with Preclinical Brain Exposure and Broad-Spectrum Potency against ALK-Resistant Mutations[J]. Journal of medicinal chemistry,2014,57 (11): 4720-4744.
2.Basit S, Ashraf Z, Lee K, et al. First macrocyclic 3rd-generation ALK inhibitor for treatment of ALK/ROS1 cancer: Clinical and designing strategy update of lorlatinib[J]. European Journal of Medicinal Chemistry,2017,134:348-356.










