
RNA测序分析有万般套路,比如
tophat+cuffLinks,star+htseq+deseq2,hisat2+stringtie
等等,但是对于这些组合得到的结果哪个更可靠,恐怕我们没有足够的精力和技术去深入研究。但是在今年七月份,一群美国人在Nature Communications上发表一篇足够有分量的文章,在精确度、效率和一致性三个层次上评估了当前主流的39个工具的120个组合,并选出了最优的工具套装。这篇文章名为《Gaining comprehensive biological insight into the
transcriptome by performing a broad-spectrum RNA-seq analysis》,下面就由小编来为大家解读下文章里关于short-read(二代测序结果)有参比对部分的内容。
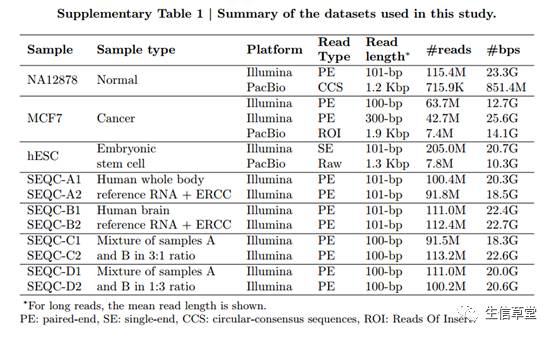
分析样本,如下表格所示,一共有15个样本,其中short-read测序样本12个,有100bp和300bp两种测序结果。
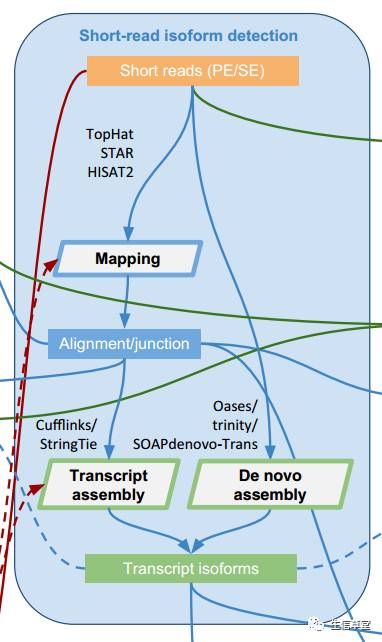
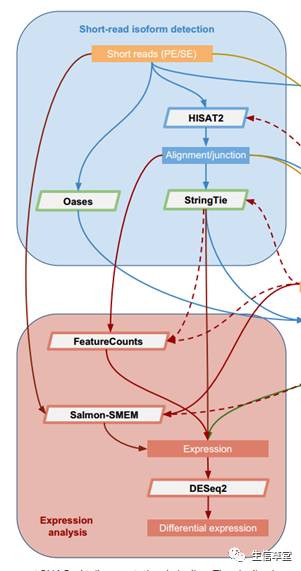
作者使用不同的软件在回帖、组装、定量以及差异计算方面分别作了测试,如下图流程所示:
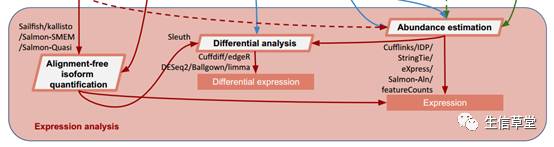
在回帖软件方面,作者主要选择了
ToHhat、STAR
和
HISAT2
这三个最流行的软件以及
RASER
。
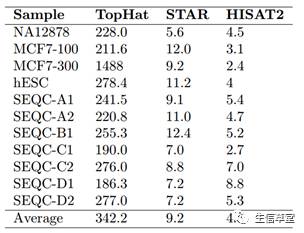
软件速度如下表所示,数值的单位为小时。
HISAT2
的回帖速度最快,其次是
STAR
,最慢的是
TopHat
,和前两者相比
TopHat
的速度是让人无法忍受的。
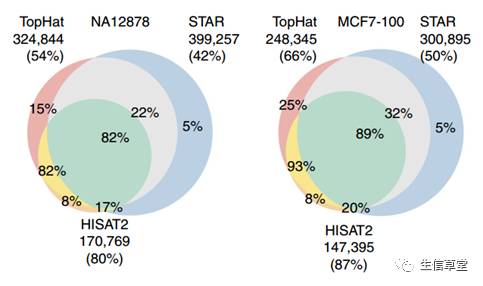
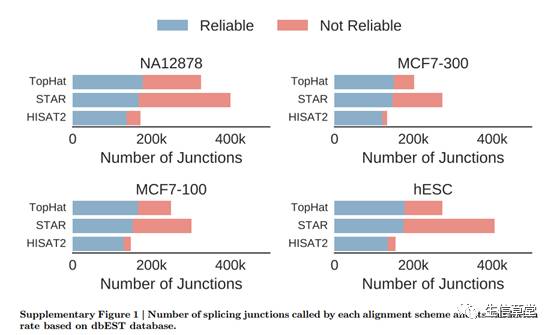
对回帖的结果进行分析,在junctions评估方面,我们可以看到
TopHat
和
STAR
虽然在数量上高于
HISAT2
,但是
HISAT2
的自己特有的junctions却是最少的。将junctions放在dbEST database检验可信度,发现
HISAT2
有最高的表现达到了80%,通过两步法mapping的
STAR
虽然得到的junctions数量众多,但是其可信的junctions比例却是最低的。
作者选取了两个最常见的软件
Cufflinks
和
StringTie
进行组装(这里针对有参,无参组装小编这里就不讲述了),从速度上看,StringTie比
Cufflinks
要快很多,其中
Cufflinks+STAR
这对组合是最慢的,
StringTie
和上游的三个软件的组合在速度方非常接近。
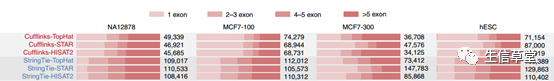
在转录本的组装数目方面,
StringTie
组装的转录本比
Cufflinks
得到的转录本在数量量多出近一倍。在100bp长度read组装方面,三个mapping软件对两个组装软件结果数量的影响相对于在300bp样本(又数第二列)下的影响小很多。
红色是敏感度,蓝色是精准度,可以发现在Gene层面上,
Cufflinks
是稍微优于
StringTie
的,但是在Transcipt层面上,
StringTie
比
Cufflinks
无论是敏感度和准确度上都是大幅领先的。有个例外就是300bp长度read组装上,
StringTie
并没有表现出在100bp read组装上的优势。

考虑到目前常规的测序长度为150bp,所以StringTie是一个更好的选择。
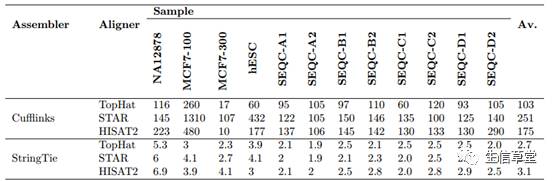
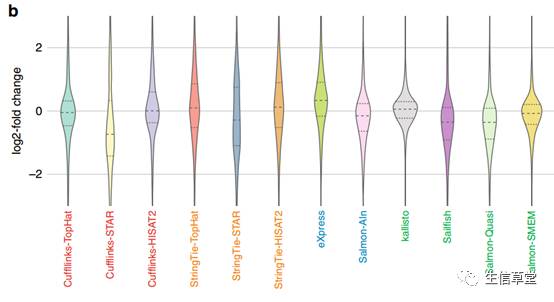
在转录本定量方面,作者既测试了
StringTie
和
Cufflinks
自带的定量结果,又加入了其他定量软件,如下表所示:
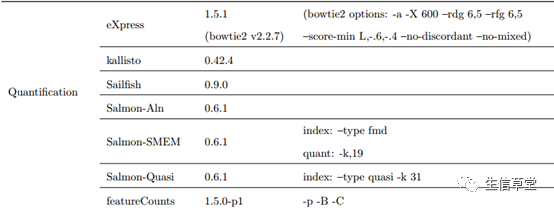
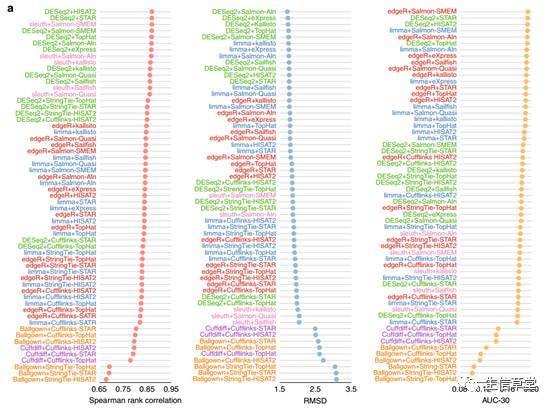
在Spearman rank correlation的热聚图上,
StringTie
相对于
Cufflink
s
与其他软件的一致性更好一点。
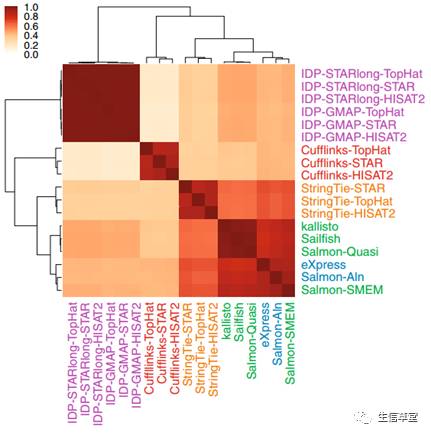
针对同一组织两个不同测序
长度
MCF7-100
(
100bp
)和
MCF7-300
(
300bp
)样本的定量结果进行分析发现,
STAR
作为回帖软件得到的结果在两个测序长度下的表达量计算结果(左
2
和左
5
)并不稳定。
kallisto
和
Salmon-SMEM
的一致性最好,
cufflinks
在一致性上稍微优于
StringTie
,但转录本数量远少于
StringTie
(参考第四部分),
HISAT2
和
TopHat
优于
STAR
。
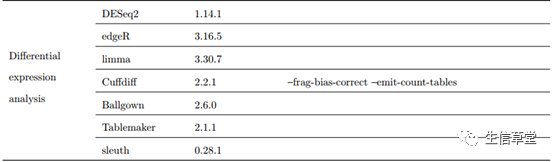
在差异计算软件的挑选上,作者除了使用
Cufflinks
套装自带的软件
Cuffdiff
外,还使用了下表所示软件。
通过三个方面的分析发现
DESeq2
的结果一致性是最好的,另外
edgeR
和
limma
略逊于
DESeq2
。
Ballgown
和
Cuffdiff
的表现让人很失望。
作者最终得到的最优选择如下图所示,回帖用HISAT2,组装和定量用
StingTie
,差异计算选择
DESeq2
。
通过这篇文章我们可以发现,不同的mapping软件得到的结果差异还是很大的,在junctions精确度上HISAT表现最优。虽然在定量上
Cufflinks
并不逊色于
StringTie
,但在组装上
Cufflinks
相对于
StringTie
在转录本数量上的弱势是很明显的,并且StrignTie的速度相比
cufflinks
要快一些。原本与
StringTie
搭配的差异计算R包
Ballgown
表现并不尽人意,
DESeq2
有着最好的差异计算表现,可以搭配
StringTie
和
HISAT
组成我们RNA分析的首选套餐。
这篇文章除了以上的分析优化外,还做了
SNP
分析优化、
long-read
流程优化和无参组装优化,这里小编就不一一说了,有需要同学可以找这篇文章仔细分析下。
在看了这篇文章后,我终于可以放心的选择合适的软件搭配了
。
生信草堂
将会与更多的优秀微信公众号合作,把最优秀的微信推文呈现给大家,希望可以帮助读者更多的了解生信技术,培养和提高读者的生信分析能力!
号外,号外,号外
你想和生信分析大神做好朋友么?
你想认识更多爱好
生信分析的小伙伴么?
你想让自己的生信分析走上快车道么?
那就赶快加入我们的微信群吧:
生信草堂交流群
或者加我们的微信,我们会把您拉入我们的社区:mly-1800; Edison686868






