慢病毒载体(LV)在获得性及遗传性疾病的基因治疗中的应用越来越广泛。这篇综述介绍了生产这些载体最前沿的技术,尤其是涉及到临床应用的大规模生产技术。相比较于在稳定细胞系生产的逆转录病毒载体,临床级别的慢病毒大多是通过在细胞工厂瞬时转染293或293T细胞生产的。然而,最近的进展倾向于采用中空纤维管反应器、悬浮培养及改进的稳定细胞系生产。正如生物技术行业的惯例一样,慢病毒生产已经建立了比较复杂的下有处理规程,包括去除任何过程来源的污染如质粒、宿主细胞DNA或蛋白。这篇综述比较了已发表的慢病毒大规模生产和纯化工艺并介绍了它们的优缺点。此外,稳定细胞系领域的进展及它们作为临床材料生产载体的进展也一并介绍。
随着欧洲第一个批准AAV1载体用于治疗脂蛋白脂酶缺乏(Glybera),基于病毒载体的基因治疗将会越来越多地应用到罕见、获得性疾病的治疗。根据治疗的目的及靶向细胞或组织的不同,会优先选择一种质粒系统。如果应用在分裂的细胞或组织中,为了达到转基因的长效表达一般需要用整合型载体。传统上,逆转录病毒载体是很好的选择,因为它们可以将基因稳定整合到细胞中。目前开发的逆转录病毒载体系统主要有两种:来源于猫白血病病毒的γ-逆转录病毒(MLV)和主要来源于HIV-1的慢病毒载体(LV)。过去很多成功的临床实验采用的是基于MLV的载体,虽然现在这个载体还在用,但是趋势是倾向于采用慢病毒载体。这种转变的原因由很多:(i)与γ逆转录病毒相比,慢病毒因为可以跨过细胞膜所以可以转染非分裂细胞。(ii)慢病毒的整合模式有别于MLV载体,在引入插入突变方面似乎比MLV安全。(iii)慢病毒可以获得比较高的滴度。
这些就是逐渐由MLV转向LV的主要原因,只是目前LV载体整体的生产条件还没有达到最大潜能及MLV载体的水平。
LV载体已经成功地应用于临床实验,尤其是免疫缺陷、神经退行性贮藏疾病等罕见病的治疗。
然而,LV在治疗遗传或获得性疾病包括β-地中海贫血、帕金森氏症及用于治疗癌症的基于嵌合抗原受体的免疫治疗在临床已经获得评价并获得了令人惊喜的结果。这意味着,鉴于这些新疗法的逐渐变为常规使用,其生产技术成为一个关键问题。
基于发表的公共信息资源,这篇综述介绍了LV载体生产的实际水平、实际操作规程及仪器设备的优缺点、所能达到的最高的生产水平,最后对以后的发展进行了展望。
LV载体系统的原型是已经深入研究的人类病毒,HIV-1。除了HIV-1之外,其它慢病毒也被作为基因转移载体(TV)来开发,但是它们绝大多数没有达到临床研究的水平,例如HIV-2、猿免疫缺陷病毒、非灵长类慢病毒包括猫免疫缺陷病毒、牛免疫缺陷病毒、山羊关节炎脑炎病毒。只有基于马传染性贫血病毒的载体被开发到临床应用。
下面这篇综述将聚焦在基于HIV-1的LV载体。
主要是考虑到HIV-1对人的病原性及相关的安全考虑,研究人员开发了不同代的LV载体系统,其中目前第三代被广泛应用在研发及临床中。它是一个4质粒系统,包括三个辅助质粒和一个转移质粒。辅助质粒的选择取决于对分裂基因组进行包装合理设计,参见Dull的描述
21
。这个生产系统与临床安全应用的所有必需特性相关联(采用非重叠的、分裂基因组包装载体来最大程度地减少潜在的重组事件,这种重组事件可能导致有复制能力的病毒的产生)。HIV-1所有非必需基因(vif,vpr,vpu,nef)仅仅在第一代载体中保留,第三代慢病毒载体已经将它们去除。与此类似,在第二代慢病毒载体中保留的调节性的tat基因在第三代中被去除,因为它的反式激活功能不是必需的,因为转移载体中5’长末端重复中的U3启动子已经被一个组成型激活的启动子序列所替代,如巨细胞病毒启动子、劳氏肉瘤病毒启动子加可选择的增强子或者可诱导/抑制的启动子序列,如7tetO
26
。这通常被说成是pRRL设计(转移载体包含嵌合劳氏肉瘤病毒,(RSV)-HIV5’LTRs)或者pCCL设计((CMV)-HIV5’LTR)。这些修改产生了需要辅助质粒的慢病毒系统,这些辅助质粒是基于来源于HIV-1的gag-pol(编码结构蛋白和病毒酶)和rev(编码翻译后调控元件)及env。虽然慢病毒载体可以用不同的异源膜糖蛋白包裹成假病毒,但是所有大规模(临床级)制备的载体所用的糖蛋白都是水泡性口炎病毒膜蛋白(VSV-g),这是因为它在下游处理时稳定性高以及比较广的转染谱。
转移质粒是唯一转移进靶细胞的遗传物质,包括待转的基因表达盒、包装、逆转录和整合所必需的顺式作用原件。考虑到提高生物安全性,自我失活的慢病毒载体也被开发出来,在这个载体中,3’LTR中的U3元件引入了缺失突变。这种载体一旦被转入靶细胞就丧失了病毒LTR的转录活性,从而使得产生有复制能力的重组子的风险最小化,同时也避免了启动子干涉相关的问题。
至于载体的生产,将四个分别编码gag-pol、rev、VSV-g及带有转基因及内部启动子的自激活型慢病毒转移质粒共转染HEK293或HEK293T(见下面)(Figure 1)。
慢病毒载体其它方面的安全性提高还有去除载体基因组中的同源序列,因为包装信号(Ψ)通过密码子优化延伸到了gag基因的第一个碱基及gag-pol载体。密码子优化使得gag-pol蛋白的翻译不再依赖于rev,因此Rev反应元件(RRE)序列可以从包装载体中去除。因为转移载体中含有部分gag序列,对gag-pol序列的密码子优化可以使得Ψ-gag重组(例如Sastry所报道
31
)变得不可能。密码子优化的另一个优点是可以使潜在的重组概率降低100倍。
转移载体中缺失RRE序列的完全不依赖rev的慢病毒载体系统目前还没有出现。RRE(未拼接的载体转录本的一部分)存在于多个Rev分子组装的寡聚复合体,该复合体可以稳定并介导载体转录本在载体生产时而不是转移载体在靶细胞整合之后从细胞核到细胞浆转运。可选择的转运序列如组成型运输元件或RNA运输元件可以在gag-pol生产时发挥作用,但是尚未证实在转移载体中与RRE一样高效。因此,目前慢病毒载体仍然利用Rev/RRE来高效的将载体基因组转移到细胞浆。
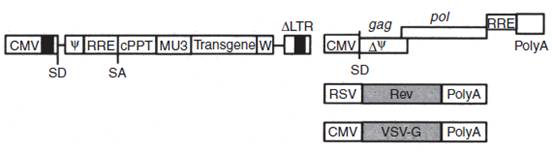
Figure 1.一个Tat非依赖的第三代HIV-1载体系统示例。上图所示的SIN转移载体含有的cPPT可以高效核转运,MSCV LTR启动子作为内部启动子及WPRE元件驱动转基因高效表达。另外三个质粒分别编码HIV-1 gag-pol、rev蛋白及VSV-g膜糖蛋白。SIN:self inactivating, 自失活;VSV-g:水泡性口炎病毒膜蛋白。
一种rev非依赖的载体技术仅仅在EIAV系统中成功实现,该系统仅需要3个质粒(两个辅助质粒gag-pol和env,还有转移质粒)。
过去,Virxsys开发了一个两质粒系统来治疗HIV感染,他是利用条件复制的慢病毒载体。这个系统的特点是将所有的辅助功能(gag-pol, rev, tat, VSV-g)都组装在一个质粒上。由于采用了全长的LTR,该系统依赖于tat因此被定义为第二代慢病毒载体系统。维持完整的LTR是为了让靶向HIV包膜基因的反义链的转录仅在HIV感染并表达tat的靶细胞中发生。虽然该生产系统在应用中易于生产、成本低廉并且比三质粒或四质粒系统产生的病毒滴度高,但是所有的辅助基因都位于一个载体上可能会产生有复制能力的慢病毒。然而,所有上述质粒组合、及转染的细胞产物中都没有发现有复制能力的慢病毒。
目前大多数慢病毒载体的生产方法都含有共转染HEK293或HEK293T细胞。优先采用HEK293T的原因是SV40 T抗原的存在可以使得慢病毒的产生更高效。此外,HEK293T的增殖能力和转染效率都要比HEK293高。目前还不能对其完整的分子机制给出解释,但是Gama-Norton等发现在基于HEK293的稳定生产细胞系中,含有SV40 T抗原的克隆产生慢病毒的能力要比不含SV40 T抗原的克隆高。这些克隆的唯一区别就是表达/不表达SV40 T抗原。Smith和Shioda也在CV-1细胞(SV40 T抗原阴性)和COS-1细胞(SV40 T抗原阳性)中证实了SV40 T抗原在慢病毒载体生产中的积极作用。COS-1主要用来慢病毒载体及小规模自动生产的筛选,因为它的载体质量(感染效率提高,细胞蛋白来源的污染少)比HEK293T高。但是,采用COS-1细胞的最大的缺点是它是完全贴壁细胞,而HEK293T细胞可以在无血清培养基中悬浮生长,这对大规模载体生产非常有利。
下面这部分比较了文献报道的实验室规模和工业化生产规模的慢病毒的生产过程。这个比较主要集中在生产者的技术方法上。讨论每种方法的性能和产率是没有意义的,因为慢病毒系统、转的基因及滴度测定方法的差异导致无法进行客观比较。但是,在这篇综述中,作者报道的滴度作为衡量指标。
以研发为目的的小规模生产是采用生长在培养皿、培养瓶、多盘系统(Cell Factories, Cell Stacks)或HYPERFlask的贴壁细胞。采用传统的磷酸钙转染或者最近发展起来的PEI转染来转染最佳密度的细胞(<50%),后者的优点是不依赖培养条件、转然后不需要换液及可用于悬浮细胞转染。原则上,小规模生产也可以采用阳离子转染试剂如lipofectamine、fugene及293fectin,它们转染也都获得了高水平表达。
通过比较不同的细胞培养系统,Aububel等并没有发现所评估的培养系统(培养瓶,一层或十层细胞工厂)所产生的慢病毒滴度有任何差异。利用HYPERFask,Kutner等发现采用HYPERFask培养可以比用传统培养装置培养所产生的慢病毒每个表面单位高10倍,原因可能是前者有更高的氧气利用率。此外,HYSPERFask生产的慢病毒比传统培养皿生产的慢病毒所含细胞蛋白、核酸污染更低。
HEK293T细胞贴壁不牢,所以采用转瓶培养生产慢病毒时更困难,同时为了要保证细胞贴壁,转染条件的优化要更加小心。在这种背景下,Patel等证实在HEK293中过表达alpha-v和beta-3整合素可以提高细胞贴壁能力,进而提高转瓶中慢病毒的产率。然而,这种方法需要采用过表达整合素的重组HEK293。
当转向临床实验时会需要大量的慢病毒载体,而将生产工艺放大则显得极为重要。这既可以通过扩展方法(增加生产单元),也可以通过采用悬浮培养系统实现,后者比贴壁细胞更容易放大。两种方法下面都会介绍:
采用贴壁细胞大规模生产慢病毒
大多数大规模生产都是小规模生产的直接放大,即通过增加培养/生产单元来实现。生产基本上采用大量的多层培养系统(Cell Factories(CF)(CF-10)(Figure 2))或者Cell Stacks(CS)。因为易于操作,10叠层装置(CS-10)是首选,虽然原则上40叠层装置同样也可以用,但是因为它重量增加的原因所以需要特殊的处理系统。此外,每层平板的气体交换及培养基层不可能一致,因此用显微镜控制40叠层装置细胞的生长很困难。生产要么采用放在层流工作台的开放模式,要么采用半封闭模式,后者对操作人员、环境及终产品的安全性更高。

Figure2.Nunc公司的十叠层细胞工厂(CF-10)
收获是通过简单的培养基置换完成的,在某些情况下,通过增加收获次数来提高最终慢病毒的质量。然而,在临床前及临床级大规模生产时,频繁收获是不现实的,因此在大多数时候只是收获1-3次。
根据10叠层培养设备的数量及收获次数的多少,传统上采用10-24个CF-10设备一次生产周期可以收获的体积介于20-52升之间。
在多次生产周期的生产规程中(多次亚批次系统),可以获得大批量的药品(纯化的和瓶装产品)。Ausubel等介绍了一种大体积(>100升)生产慢病毒的方法,该方法把几周内生产的多个10升的亚批次产品收集到一起。Dupont也使用了类似的方法。单独处理每次收获的样品纯化的产品每次都要进行质控。为了生产大量的药物产品,所需数量的纯化的分装品会经过解冻、汇集、无菌过滤并装瓶。
在稳定诱导型生产细胞系(tet-off诱导系统)条件下,50-LWAVE反应器(工作体积25升)在一次连续培养过程中可以收获138L含有慢病毒的细胞培养上清(收获时间:诱导后的8天内收获3-6次)(见下面)。
未处理的上清一般含有1-5×10
7
个感染颗粒/毫升,与报道的小规模生产的滴度相仿。然而,Greene等及Ausubel等也分别报道采用大规模生产获得了0.5-1×10
7
及0.5-2×10
6
转染单位/毫升的病毒滴度。这些差异主要是受载体组成如目的基因、所用的启动子及其它的调控元件,同时也受滴定方法的影响,因为目前滴定方法还没有标准化。
所有的大规模生产规程都是针对HIV-1及EIAV慢病毒载体开发的。
最近报道了一种采用中空纤维的慢病毒生产系统,被称为准‘大规模’。首先将HEK293T接种到中控纤维中,贴壁24小时之后将三个质粒转染进HEK293T细胞。它的优点是它是封闭的全自动培养系统,并且产量与三个CF-10叠层相当。但是在实际生产中它需要建立多个平行系统。
Table 1罗列了可利用的大规模慢病毒生产体系。
虽然转染贴壁细胞是生产慢病毒的金标准方法,但是这个方法在扩大规模上受限。对工业化生产来说,在大的生物反应器中培养细胞通常是最便捷的方式。
在生物反应器中生产需要对悬浮的生产细胞扩大培养。多个用于生产慢病毒的细胞系(293T、293FT、293SF-3F6)被报道易于在化学成分限定的培养基(Freestyle 293 and F17, Invitrogen, Carlsbad, CA; HyQSFM4TransFx293, Hyclone, Logan, UT)中适应悬浮培养。这些细胞可以在没有为载体的情况下迅速悬浮生长,这使得它们的培养和扩大比贴壁培养的细胞容易很多。此外,培养基中没有牛血清及其他动物来源的组分是临床生产最合适的情况,它可以降低被外源物质污染的风险。
在悬浮培养模式下,细胞可以通过不同的容器进行扩大:摇瓶、玻璃生物反应器、不锈钢生物反应器、培养袋及一次性搅拌容器。有报道采用HEK293T细胞进行扩增、转染、慢病毒生产可以用一次性的生物反应器实现50L的规模。
采用磷酸钙沉淀的DNA转染悬浮细胞由于持续的搅拌而被认为效率低下。因此,大多数时候采用其它的转染试剂如阳离子聚合物。线性25kDa的PEI可以在293T及293-EBNA1中诱导最高的转染效率,用GFP质粒作为报告质粒可以达到75%的转染效率。在Marceau及Gasmi的关于生产慢病毒的专利中也采用了同样类型的PEI,他们达到了90%的GFP阳性细胞。然而在这个例子中,转染效率是在转染后48小时检测的,因此GFP信号可能来自新合成的慢病毒的感染。
为了在几个HEK293来源的细胞系中获得最佳的转染效率,细胞密度似乎是一个很重要的参数。许多文章一致认为PEI/DNA聚合物转染前细胞的最佳密度是1百万细胞/毫升,更精确点是介于8×10
5
细胞/毫升至1.5×10
6
细胞/毫升之间。
PEI转染的一个缺点是达到高转染效率所需要的质粒DNA的量。Ansorge等1ug/10
6
293SF-3F9细胞,而Marceau及Gasmi报道最佳的用量是2.5ug/10
6
293T细,这样在大规模制备时会需要大量的质粒(例如,一个200L的生物反应器需要750mg质粒DNA)。这么大量的DNA意味着原料成本过高,大量残留DNA需要在后续处理中去除。
PEI的另一个问题是在生产或纯化慢病毒时缺少检测及定量PEI的方法。因此,还不清楚PEI是否会和慢病毒共纯化,这种共纯化的水平及是否会对慢病毒的感染能力及稳定性产生破坏作用。
另外,有报道证实电穿孔可作为真核细胞的转染方法。Witting等报道采用电转可以高效的生产慢病毒。然而,这种方法需要在电转时将细胞浓缩到10
8
/毫升,因此很难扩大到工业规模。事实上,现在的方法是在电转前浓缩,电转后稀释到原来的体积。更大规模的操作需要例如连续离心机等特殊设备。一般来说,细胞培养阶段的技术操作控制地越少越好,因为他们会增加微生物污染的危险。此外,离心也可能会导致细胞压力及大量细胞损伤,进而会降低病毒产量。因此,虽然很有前景,但是电转要想在工业规模获得应用还需要进一个完善和简化。
在前面瞬时转染悬浮细胞生产慢病毒的例子中,培养基中加入了终浓度为1-10mmol/L的丁酸钠。像其它组蛋白去乙酰化酶抑制剂如曲古霉素A、丙戊酸一样,丁酸钠据报道可以组织DNA凝集,从而使得启动子暴露出来。这样会提高RNA转录及后续的慢病毒产量。然而,丁酸钠的作用还存在争议,因为研究者之间慢病毒的产量并不一致。Ansorge等报道采用5mmol/L的丁酸钠将VSV-g包被的慢病毒的产量提高了10倍。与此相反,Sena-Esteves等采用10mmol/L丁酸钠并没有看到VSV-g包被的慢病毒的产量有任何提高,反而在其它包被类型的慢病毒中看到了产量提高。因此,很难做出组蛋白去乙酰化酶抑制剂作用的结论。这些实验的主要区别可能来自质粒载体本身,因为它们与组蛋白的相互作用理论上依赖于DNA序列及启动子的类型。因此,丁酸钠对每种DNA载体的影响应该单独评估。公布的数据表明,在悬浮培养的生产的慢病毒滴度类似于贴壁细胞获得的滴度。在批次收获或培养上清中获得的慢病毒滴度在10
7
~10
8
IG/ml或TU/ml范围内,而贴壁细胞系统获得的滴度在10
6
-10
8
TU/ml(Table 1)。然而,这种比较是非常困难的,因为可供比较的采用相同载体的报道数量少,不同的分析手段也是一个原因。
最后,在贴壁培养系统中,培养上清可以间隔一天收获两次,这是一个提高成本效益的好方法。在悬浮培养系统中,这种方法很难实现。Ansorge评估了一种灌注系统,该系统在7天中可以实现培养基替换和连续收获。他们的结果证实转然后48-96小时之间是慢病毒滴度最高的,说明在悬浮培养系统中多次收获也是可行的。虽然灌注系统在大规模时复杂且成本高,但是他对瞬时转染具有巨大吸引力。以为它可以在保持质粒用量不变的情况下,可以将收获体积扩大2-3倍。
总之,利用悬浮细胞瞬时转染大规模生产慢病毒是可行的,并且显示出良好的产量。但是,在转移到工业生产之前,该技术还需要进一步完善。优化的主要瓶颈是转染过程本身,它需要大量质粒DNA,从而使生产过程极其昂贵。一种工业友好、减少DNA的消耗、提高生产细胞的百分比的转染技术是使这一过程在未来工业中有利可图的关键因素。
如果没有展望中提到的改进,一旦用于慢病毒生产,稳定的生产细胞系将代表一个负担得起的生产系统。
相较于瞬时转染生产,稳定的慢病毒生产才是基因治疗的最佳选择。后者可以降低生产成本、提高整体安全性和可重复性,这也是限制瞬时转染技术大规模应用的主要障碍。首先为临床实验、最后为基因治疗产品商业化实现稳定的生产系统是降低生产成本、提高慢病毒质量和安全性的重要里程碑。自上世纪90年代初第一个包装细胞产生时,就发展出了很多构建策略,这些策略的主要差别在于选择导入编码基因的载体的方法。例如,质粒和整合载体以及所利用的包膜蛋白。后者已成为导致更多诱导性而非组成型包装细胞发展的最重要因素之一。VSV-g作为应用最广泛的慢病毒异源包膜蛋白,它有很强的细胞毒性。它的表达必须在慢病毒生产时诱导表达来防止包装细胞死亡。此外,HIV的gag和pol基因的表达也有细胞毒性和抑制细胞生长活性,因此这些基因的高表达必须控制在病毒生产阶段。第一个常用的诱导系统是Tet-on和Tet-off系统,它通过向培养基中添加或去除四环素/多西环素来作用于四环素反应元件(TRE)来诱导基因表达。当其它适合慢病毒包装的非毒性包膜蛋白被发现时,随后会开发出组成型包装细胞。这部分将分别讨论来源于HIV-1和EIAV的诱导型和组成型包装细胞,它们的性能指标总结在Table 2中。
Tet-off
诱导系统。
四环素诱导表达系统在第一代包装细胞中就有应用,该系统中HIV gp120/gp41包膜蛋白还没有被VSV-g替换掉,调节性及非必需基因仍然保留在包装载体中。Hela来源的HtTA-1细胞组成型表达tTA,它是有大肠杆菌的四环素抑制子和单纯疱疹病毒的HSV-VP16反式激活因子融合的一个嵌合蛋白。tTA一方面控制所有包装基因的表达,另一方面也控制调节基因Rev蛋白的表达,而Rev会激活gag和pol基因。随后,Tet-off系统被用在基于HEK293的稳定包装细胞系SODk1中调控VSV-g包膜蛋白和第一代包装载体,该系统可以3-4天内生产滴度超过10
6
TU/ml的病毒颗粒。随后两个独立的课题组构建了基于多西霉素抑制HIV-1 Rev/Gag/Pol及VSV-g包膜蛋白的第二代稳定细胞系,它们生产的慢病毒滴度在3.5-5×10
6
TU/ml。此外,Tet-off系统也被用于在SODk1细胞中表达条件型SIN慢病毒,随后用于SODk3第三代包装细胞系中,该系统去除了HIV-1Tat反式激活蛋白及除了Rev之外的非必需基因。在这个cSIN转移载体系统中,U3转录调控元件被Tet反应元件TRE替代。这种载体设计保留了SIN的特征,同时只能在表达Tet调控的反式激活因子tTA的细胞中生产慢病毒。标准的SIV转移载体必须要通过质粒转染整合到稳定包装细胞中来避免逆转录后5’LTR失活,与此相反,cSIN转移载体可以通过转导进入细胞。事实上,逆转录发生之后,在多西霉素存在下,TRE会引导载体中含有包装信号的前病毒全长基因组转录来进行慢病毒包装。Tet调控的SODk1和SODk3包装细胞转导cSIN转移载体之后,会分别生产出高浓度(>106TU/ml和1×107TU/ml)的cSIN重组载体。
发展第二代包装细胞的另一种策略是Virxsys建立的抗HIV基因治疗,该方法是在293细胞中采用三水平级联基因调控系统来避免毒性,包装、包膜蛋白调控的基因的渗漏表达,组成型表达的tTA的毒性。此外,对Tat、Rev、Gag-Pol基因的密码子进行了优化来减少同源重组的发生。这种新方法虽然降低了包装基因和VSV-g的总体毒性,但是它仍然不能抑制p24Gag的渗漏表达。含有抗HIV包膜蛋白基因反义核酸的VRX496载体可以在超过11天的时间内生产出最高浓度相当于3.5×10
7
TU/ml和p24Gag 300ng/ml的慢病毒。并没有检测到有复制能力的慢病毒,但是长期分析发现培养2-3个月之后有部分基因沉默。Virxsys及其它讨论的包装细胞是采用质粒构建的,这些质粒随着时间推移经常会发生沉默,这也可以解释慢病毒的短期稳定性。为了克服这些问题,2003年Ikeda及2009年Throm报道了采用整合型MLV载体作为整合型载体基因的导入工具。St.Jude用于治疗X连锁重症联合免疫缺陷疾病的技术是基于构建可诱导的第三代293T来源的GPR(gag-pol-rev)和第二代GPRT(gag-pol-rev and tat),在上述系统中,Rev和Tat的表达受Tet-off系统紧密调控。高表达的Rev基因会诱导其它的包装基因、VSV-g基因和SIN转移载体CL204i-EF1α-hγc-OPT,后者表达密码子优化的IL-2Rγc的cDNA,并且其两端有400bp鸡β球蛋白HS4的绝缘子序列。Ikeda报道的原始方法利用LTR驱动MLV,而Throm及其合作者应用SIN-MLV载体来避免LTR-MLV包装基因组被包装进慢病毒颗粒。该方法引入的另一个创新点是为连接有抗生素抗性基因的SIN载体基因组整合而开发的连环阵转染技术。表达GFP或IL2RG基因的生产细胞可以生产出滴度超过5×10
7
TU/ml的上清。如前所述,按照支持X连锁重症联合免疫缺陷疾病临床试验的规模分两次生产的大约280L CL204i-EF1α-hγc-OPT慢病毒,经浓缩之后,其滴度达到4.5和7.2×10
8
TU/ml。
St.Jude儿童研究医院利用GPRGT包装细胞及表达Wiskott-Aldrich综合症蛋白的650MNDhWASp1包装细胞生产临床级别的VSV-g包被的Wiskott-Aldrich综合症慢病毒。为了获得生产细胞,将U3区含有HS4染色质绝缘子的转移载体采用一种改进的方法导入到细胞,该方法是在Throm的基础上改进的。将hWASp 单体连接到博来霉素抗性基因而不是更长的连环阵列稳定转染GPRGT细胞。该细胞克隆可以生产滴度超过1×107TU/ml未浓缩的慢病毒,并且在连续传代时可以稳定生产超过8星期。
据我所知,到目前为止,只有GPRG-EF1α-hγcOPT包装细胞已经用于临床试验,而650MNDhWASp1细胞仅仅打算最近用于临床试验,该研究是由St.Jude儿童研究医院开展的。
最近,第一个生产整合型缺陷慢病毒载体的稳定包装细胞由Tal Kafri小组开发出来。该包装细胞是通过转染含有整合酶D64E突变体的四环素调控的包装载体pTK1574和VSV-g基因进入PVG3细胞,该细胞稳定表达反式激活因子tTA。生产细胞的建立要么通过转导cSIN转移载体(滴度:10
7
TU/ml),要么通过稳定转染一个新的多聚嘌呤位点缺失的转移载体(滴度:10
8
TU/ml)。这两种整合缺失的慢病毒载体都适合体内转导大鼠纹状体中的神经元。
Tet-on
诱导系统。
Oxford BioMedica在293T细胞表达的EIAV慢病毒的基础上开发了一种稳定包装细胞系和一个Tet-on诱导系统,在该系统中,向培养基中添加多西霉素之后,Tet抑制子(TetR)紧密调控VSV-g和Gag/Pol的表达。该系统与Tet-off系统相比渗漏表达更低。EIAV转移载体编码一个治疗帕金森氏症的基因治疗产品:ProSavin。两种ProSavin生产细胞PS5.8和PS46.2被详细鉴定,它们即使在没有选择压力的情况下也可以稳定培养49天。病毒的滴度平均小于10
6
TU/ml,与瞬时生产系统相当。
Tet-on/cumate
诱导系统。
Tet-off诱导系统不能完全消除VSV-g的渗漏表达从而导致细胞毒性及包装细胞不稳定,因此开发了一种叫293SFPacLV的双开关系统。该细胞来源于在无血清悬浮培养基中生产慢病毒的293SF细胞。293SFPacLV细胞表达cumate开关系统的CymR抑制子及Tet开关系统的反式激活因子rtTA2S-M2。基因诱导是通过向培养基中添加多西霉素和cumate诱导剂来实现的。在转导表达GFP的cSIN慢病毒到包装细胞之后,生产细胞可以产出3.4×10
7
TU/ml的慢病毒。然而,尽管GFP标记基因获得了令人鼓舞的结果,但这些细胞从未进入临床研究阶段。
蜕皮激素诱导系统。
该系统是基于昆虫蜕皮激素类似物蜕皮激素A,被用于四环素调控技术的替代技术。蜕皮激素A反应性293T细胞系中,gag、pol、rev和VSV-g的表达是由诱导型蜕皮激素启动子的控制之下。这种细胞不断产生二代慢病毒,经浓缩后可以达到10
8
TU/ml。
与此类似,293-Rev/Gag/Pol细胞系是将HIV rev和gag/pol基因分别置于独立的蜕皮激素诱导型启动子下,然后导入293细胞。在筛选时,该细胞持续加入HIV特定的蛋白酶抑制剂 Saquinavir来控制HIV蛋白酶的细胞毒性。诱导后48小时之内,293-Rev/Gag/Pol细胞释放出大量的HIV Gag/Pol颗粒(大约10ug p24/ml),它可以将第三代HIV载体包装到很高的滴度。
毫无疑问,具有高产量的组成型(或连续型)包装细胞的获得要比诱导型细胞难获得的多。事实上,载体基因的毒性使得VSV-g包膜蛋白或筛选p24Gag高表达蛋白变得不可行。因此,这种类型包含仅仅带有不是VSV-g的包膜蛋白的包装细胞,它们与诱导型细胞比起来通常产生比较低的p24Gag。第一个名叫STAR的连续型包装细胞系是由Ikeda于2003年开发出来的。STAR引入了多方面创新:(i)测试了三种gamma逆转录病毒的包膜蛋白:R-多肽切割位点引入HIV蛋白酶位点的猫科动物的内源性γ逆转录病毒RD114包膜蛋白(RD114-Pro),带有MLV的胞质尾的长臂猿白血病病毒(GALV+),MLV 4070A (Ampho)包膜蛋白。(ii)通过MLV整合载体而不是质粒稳定转染来导入包装基因。(iii)除了293T细胞,还测试了Hela和HT1080细胞。Gag-pol基因在MLV LTR的转录调控之下,而其它的载体基因通过标准质粒表达。STAR技术适用于生产第二代和第三代慢病毒,可以连续3个月生产出最高到850ng/ml的p24,其浓度达到10
7
TU/ml。然而,没有STAR来源的生产细胞到达临床应用,因为它可能将编码Gag-Pol和Rev基因的MLV基因组错误包装到慢病毒颗粒。随后发展的WinPac细胞中的SIN-MLV载体联合重组酶介导的表达盒交换技术(RMCE)降低了这种可能性。RMCE首先采用整合转移载体的稳定γ逆转录病毒包装细胞来交换不同的转基因,而在WinPac细胞中,RMEC技术被用于整合包装基因。一个表达GFP标签的SIN-MLV载体用来筛选293FT细胞中最佳的表达位点,用一个靶向载体将GFP表达盒与密码子优化的HIV-1 Gag-Pol表达盒交换。虽然RMCE是一个筛选基因组中开放转录位点的非常有用的方法,但是在这里是用它似乎有点多余,因为筛选出来的支持GFP高表达的细胞不能支持具有细胞毒性的Gag-Pol基因产物的持续高表达,更重要的是,无论采用什么导入方法,最好的Gag-Pol表达位点可以非常简单直接的采用p24Gag ELISA检测细胞上清来筛选。没有毒性的包膜蛋白RD114-PR、rev基因及载体基因组质粒被顺序转染到生产细胞,然后用抗生素筛选。获得了超过10
6
TU/ml的慢病毒,经过浓缩之后可以提高到10
8
TU/ml。降低胎牛血清的浓度到1%并在HYPERFlask中扩大培养,获得了持续到第六次收获、浓度超过5×10
6
TU/ml的慢病毒。
另一种组成型包装技术是以MolMed S.p.A开发的RD-MolPack细胞为代表。RD-MolPack系统与STAR、WinPax细胞类似,也是基于HEK-293T细胞开发的,也表达RD114包膜蛋白。与WinPac的RD-114-PR包膜蛋白不同,RD-MolPack细胞带有RD114-TR包膜蛋白,该蛋白含有RD114蛋白的胞外和穿模结构域并融合有MLV-Ampho4070膜蛋白的胞质尾巴,从而利于整合入慢病毒载体。RD-MolPack细胞独一无二的地方在于,HIV-1 gag、pol、rev及潮霉素抗性基因是通过将嵌合baculo-AAV感染293T细胞导入的。细胞首先转染表达AAV Rep78的质粒来引导baculo-AAV载体整合入基因组。产生的表达两个拷贝gag-pol-rev基因的中间克隆PK-7显示出极高的遗传稳定性,它可以在有或没有潮霉素筛选的情况下连续培养一年,其生产的p24Gag水平分别为6.7±3.5和15.3±8.4ng/10
6
细胞/天。从PK-7细胞分别独立衍生出RD2-MolPack和RD3-MolPack来分别生产第二代和第三代慢病毒。与St.Jude儿童研究医院的MolMed的策略类似,包膜基因和Tat基因(仅指RD2-MolPack细胞)是通过VSV-g包被的SIN-
L
V而不是SIN-MLV整合载体导入的。在构建包装细胞中使用MLV或者HIV SIN的重大安全问题是遥远的,但现实的可能是在新产生的慢病毒颗粒中动员了载体基因(env或gag-pol)。事实上发现,SIN-LV 真载体整合时东动员频率在0.1-0.03%左右,而SIN-MLV则会有一些痕量的3’LTR启动子活性。利用特殊的实验已经排除了上述安全问题及产生有复制能力慢病毒的可能。RD2-MolPack-Chim3生产细胞,表达抗-HIV chim3治疗基因(HIV Vif的显性负性突变体),其产生的慢病毒超过了瞬时转染人脐带血造血干细胞所生产的VSV-g包被的慢病毒。从RD3-MolPack-GFP生产细胞生产的表达GFP的SIN-LV可以转染90%的人外周血淋巴细胞,其MOI=3,水平与VSV-g包被的瞬时转染获得的慢病毒相当。有意思的是,RD2-和RD3-MolPack细胞生产的慢病毒滴度(10
6
TU/ml,浓缩后10
8
TU/ml)比通过用RD114-TR包膜蛋白瞬时转染生产的慢病毒的滴度高。
上述展示的不同系统不能简单的进行比较,因为大多数情况下,细胞生产能力指标TU或ng p24/细胞/天并不是标准的分析方法。对于稳定的包装细胞,细胞生产力必须毫无疑问地考虑,这是使一个稳定的系统有价值或没有进一步发展的最重要的参数之一。低生产力的细胞可以通过慢病毒载体的高感染力来补偿,而这通常与高转导效率有关。
下游回收过程应以最低的成本为产品提供所需的浓度、纯度和其他质量属性。这一说法适用于所有大规模的制造过程,但往往不适用于研究目的的小规模净化规程。
大多数研究级别的慢病毒是通过批次浓缩而不是粗制剂之后应用的,浓缩基本上是通过两步离心法产生的。在70000g通过超速离心浓缩后的慢病毒再通过蔗糖缓冲(50000g)纯化,然后溶解在配方缓冲液中。一种改进,特别是纯度方面的改进,代表了基于离心/色谱联用的纯化方法。例如,Kutner等评估了组合纯化/浓缩方法。蔗糖缓冲的超速离心结合阴离子交换层析获得了88.2%的收率,而相反的组合则获得了77.6%的收率。在两种方法中,浓缩都超过了100倍,病毒滴度都超过了10
10
TU/ml(VSV-g包被的慢病毒)。
这些方法最大的缺点是它们无法进行放大,最后病毒的纯度由于细胞、培养基组分或者处理过程引入的污染而达不到体内应用。浓缩之后的慢病毒潜在地可能会在体内导致副作用或转导效率降低。因此,基于色谱和膜分离的技术的纯化方法被开发出来。其优点是,这样的净化方案可以以可扩展的方式开发,并且可以实现对慢病毒的工业级净化。在这一背景下,几个研究小组已经表明,基于色谱和膜分离方法的纯化方法不仅保证了载体安全性,而且显著提高了载体效能。γ逆转录病毒和慢病毒下游加工原理的总体概述已经由Segura和Rodrigues等人发表。
考虑到工业应用,在生物技术行业传统上使用的工艺步骤已经开发用于慢病毒的下游处理。它们是基于膜(过滤/澄清,利用切向流过滤进行浓缩/渗滤,基于膜的色谱)和色谱(离子交换色谱,亲和色谱,体积排阻色谱)的技术。这些不同的过程步骤的组合是可变的,在某些情况下,不同的净化原则可以用于相同的目的。此外,采用benzonase/DNase降解污染的DNA要么是是下游处理的一个步骤,要么在病毒生产阶段已经处理过。










