生信技能树的转录组学习开班了, 第一个任务是安装软件, 于是我花了一个下午时间和Linux斗智斗勇。
系统准备
windows10: Unbuntu on windows10. 至于如何win10上开启Linux子系统,百度会有无数教程的。
建议搭配cmder,界面更好看,用的更开心。
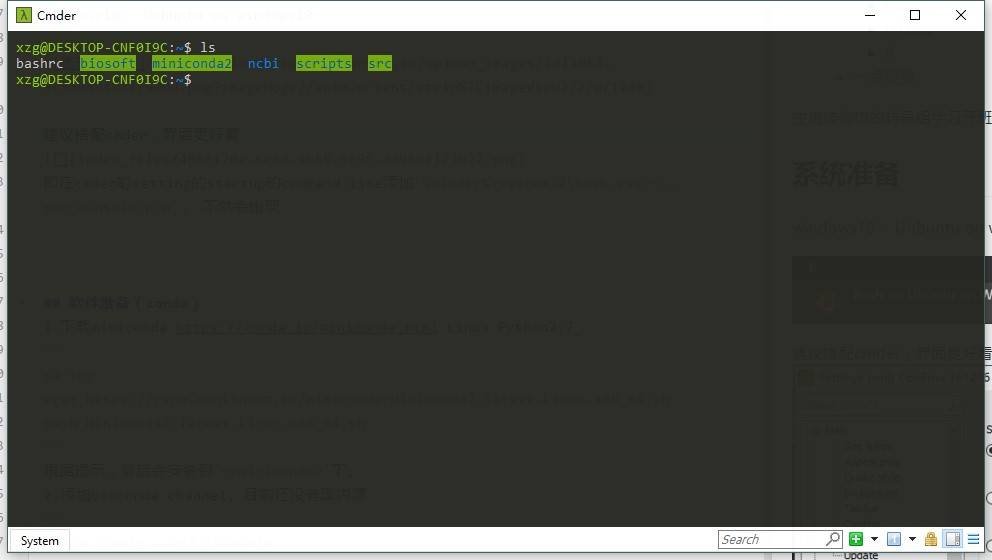
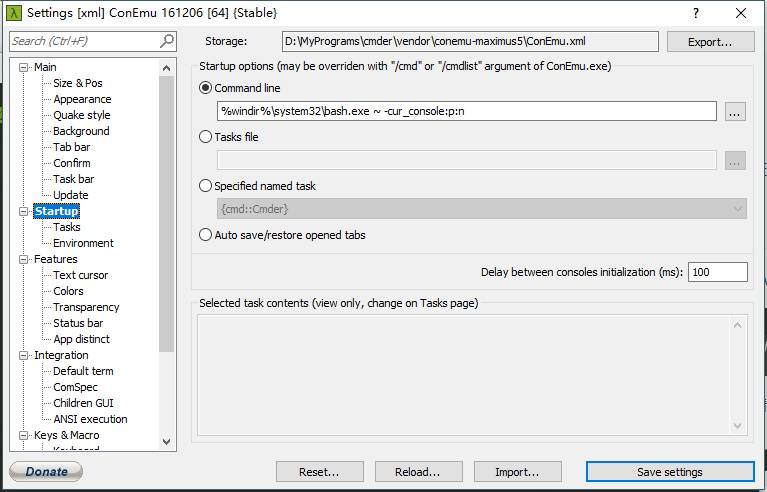
但是直接在cmder里启动ubuntu不能使用方向键,需要做一些修改,即在cmder的setting的startup的command line添加
%windir%\system32\bash.exe ~ -cur_console:p:n
软件准备(conda)
1.下载miniconda https://conda.io/miniconda.html Linux Python2.7
cd src
wget https://repo.continuum.io/miniconda/Miniconda2-latest-Linux-x86_64.sh
bash Miniconda2-latest-Linux-x86_64.sh
根据提示,最后会安装到
~/miniconda2
下。
2.添加bioconda channel, 目前还没有国内源
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/msys2/
conda config --add channels bioconda
conda config --set show_channel_urls yes
3.用conda安装软件sratoolkit,fastqc,hisat2,samtools,htseq-count, 与网络有着密切的关系
查询可供安装的软件, https://bioconda.github.io/recipes.html#recipes
conda create -n biostar sra-tools fastqc hisat2 samtools htseq
拓展: 了解conda的命令
注:conda只有一个问题,就是看网络条件,国内源似乎还在制作中。
R语言和Rstudio就看下面的讲解。
软件准备(麻烦的编译篇)
我的习惯:
-
家目录下创建src文件夹,用于存放软件包
-
家目录下创建biosoft文件夹,用于安装软件
为了提高下载速度,我们需要替换
/etc/apt/source.list
中默认镜像源。方法参考自中国科学技术大学开源镜像站
# 备份
cd /etc/apt/
sudo cp source.list source.list.bk
# 替换
sudo sed -i 's/http/https/g' sources.list
sudo sed -i 's/archive.ubuntu.com/mirrors.ustc.edu.cn/g' sources.list
sudo sed -i 's/security.ubuntu.com/mirrors.ustc.edu.cn/g' sources.list
# 更新
sudo apt-get update
sudo apt-get upgrade
选择合适的镜像站,让你的速度飞起来
sratookit
功能: 下载,操作,验证NCBI SRA中二代测序数据
网址:https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software
步骤:
cd src
wget https://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.8.2-1/sratoolkit.2.8.2-1-ubuntu64.tar.gz
tar -zxvf sratoolkit.2.8.2-1-ubuntu64.tar.gz
mv sratoolkit.2.8.2-1-ubuntu64 ~/biosoft
# 加入环境变量
echo 'PATH=$PATH:~/biosoft/sratoolkit.2.8.2-1-ubuntu64/bin' >> ~/.bashrc
# 测试
prefetch -v
# 尝试下载,默认存放在家目录下的ncbi文件夹中
prefetch -c SRR390728
阅读官方文章进一步了解:
-
如何开启ascp加速下载
-
vdb-config更改基本设置
fastqc
功能: 可视化展示二代测序数据质量
网站:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
步骤:
# 判断系统是否安装java
java -version
# 安装java, 请改成openjdk-9-jdk,下面的是错误演示
sudo apt install openjdk-9-jre-headless
# 验证
java -version
# openjdk version "9-internal"
# OpenJDK Runtime Environment (build 9-internal+0-2016-04-14-195246.buildd.src)
# OpenJDK 64-Bit Server VM (build 9-internal+0-2016-04-14-195246.buildd.src, mixed mode)
# 安装fastqc
cd src
wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.5.zip
unzip fastqc_v0.11.5.zip
mv FastQC/ ~/biosoft/
cd ~/biosoft/FastQC/
chmod 770 fastqc
# 添加环境变量, 我用sed修改
sed -i '/^PATH/s/\(.*\)/\1:~\/biosoft\/FastQC\//' ~/.bashrc
source ~/.bashrc
fastqc -v
# FastQC v0.11.5
拓展:
-
了解fastqc结果中各个图的含义
-
掌握如何从fastqc的结果中提取数据
-
学习sed的用法,http://dongweiming.github.io/sed_and_awk/
samtools
SAM: 存放高通量测序比对结果的标准格式
功能: Reading/writing/editing/indexing/viewing SAM/BAM/CRAM format
网站: http://samtools.sourceforge.net/
安装:
cd src
# prerequsite
## system requirement
sudo apt install autoconf libz-dev libbz2-dev liblzma-dev libssl-dev
### zlib2
wget http://zlib.net/zlib-1.2.11.tar.gz
tar -zxvf zlib-1.2.11.tar.gz && cd zlib-1.2.11 && make && sudo make install && cd .. && rm -rf zlib-1.2.11
### bzip2
wget http://bzip.org/1.0.6/bzip2-1.0.6.tar.gz
tar -zxvf bzip2-1.0.6.tar.gz && cd bzip2-1.0.6 && make && sudo make install && cd .. && rm -rf bzip2-1.0.6
### curses
sudo apt-get install libncurses5-dev
### htslib
git clone https://github.com/samtools/htslib.git
cd htslib
autoreconf
# building samtools
git clone https://github.com/samtools/samtools.git
cd samtools
autoconf -Wno-syntax
./configure
make && make install prefix=$HOME/biosoft/samtools
## add PATH
sed '/^PATH/s/\(.*\)/\1:~\/biosoft\/samtools\/bin/' .bashrc -i
source ~/.bashrc
samtools --help
顺便安装bcftools
cd src
git clone https://github.com/samtools/bcftools.git
make && make install prefix=$HOME/biosoft/bcftools
make clean
sed '/^PATH/s/\(.*\)/\1:~\/biosoft\/bcftools\/bin/' .bashrc -i
source ~/.bashrce
bcftools -h
因为用的是github,所以以后更新就用下面命令
cd htslib; git pull
cd ../bcftools; git pull
make clean
make
吐槽: 编译的时候需要安装好多前置包,真麻烦!
HISAT2
功能: 将测序结果比对到参考基因组上
网站: http://ccb.jhu.edu/software/hisat2/index.shtml
安装:
cd src
wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.1.0-source.zip
unzip hisat2-2.1.0-source.zip
# 编译hisat2
cd hisat2-2.1.0
make
rm -f *.h *.cpp
cd ../
mv hisat2-2.1.0 ~/biosoft/hisat2
# add to PATH
sed '/^PATH/s/\(.*\)/\1:~\/biosoft\/hisat2/' ~/.bashrc -i
source ~/.bashrc
# test
hisat2 -h
吐槽: 居然没有make install !!!
拓展:
HTSeq
功能: 根据比对结果统计基因count
# prerequsites
sudo apt-get install python-pip
pip install --upgrade pip
sudo apt-get install build-essential python2.7-dev python-numpy python-matplotlib
## 验证, 保证无报错
python -V
## python
python
>>> import numpy
>>> import matplotlib
## install HTSeq
pip install htseq
## 验证
python
>>> import HTSeq
教程:
-
http://www-huber.embl.de/users/anders/HTSeq/doc/tour.html#tour
推荐:
-
推荐安装一个ipython,学习ipython如何使用
-
将软件包安装到当前用户目录下
pip install --user xxx





