在上一篇中,本来说好的是“
如何提取拟南芥
(或非模式物种)
基因的启动子序列_基于R”
,结果却只说了提取拟南芥的情况。
这里我再补充下针对非模式物种。这里有两个策略,一个是构建对应的BSgenome包,另一个就是直接在
Biostrings
::
getSeq
()
这个函数不用BSgenome,而是用其他的支持的对象。我先说简单的,即在getSeq那边支持。
我直接在网页上搜索了,然后看到了这个回答
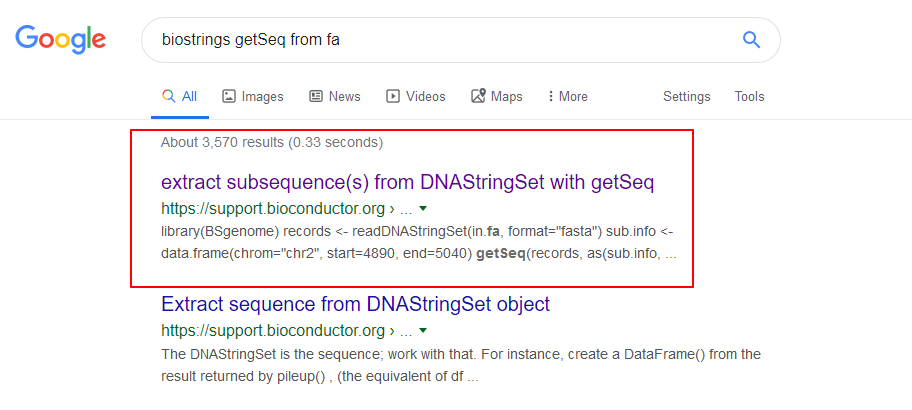
参考了这个回答,就可以直接用fa来提取啦。结果跟BSgenome是一样的。
> records readDNAStringSet("~/reference/genome/TAIR10/Athaliana.fa", format="fasta")
> promoter_gtf_part_seq getSeq(records,promoter_gtf_part)
> promoter_gtf_part_seq
A DNAStringSet instance of length 2
width seq names
[1] 4130 CCCTAAACCCTAAACCCTAAACCCTAAACCTCTGAATCCTTAATCCC...GGTTTCTGGTAAATGGAAGCTTACCGGAGAATCTGTTGAGGTCAAGG AT1G01010
[2] 4500 ATCCGCAACAATTCACCAATTGAAGAACAAGAGAAAGGTTTAAACTT...GAGAGAGAGCAATGGCGGCGAGTGAACACAGATGCGTGGGATGTGGT AT1G01020
writeXStringSet(promoter_gtf_part_seq,
filepath = "test.fasta",
format = "fasta")
至于BSgenome的构建,看起来不太划算,但实际上BSgenome还支持其他的一些骚操作,比如某些包就是基于BSgenome来找motif,所以搞一个还是比较划算的。我这里基于的构建方法是How to forge a BSgenome data package。构建还是比较简单的。就是你要创一个文件夹,文件夹里面有fa和seed。
唯一的问题在于BSgenome的fa传进去的时候
染色体一定要是分开的,Chr1.fa……ChrM.fa
。不能直接传一个genome.fa进去。(这里卡了好久……)
The sequence data must be in a single twoBit file (e.g.musFur1.2bit) or in a collection of FASTA files(possibly gzip-compressed)
所以我们先把染色体分割了。
awk '/^>Chr/ {OUT=substr($0,2) ".fa"}; OUT {print >OUT}' Athaliana.fa
这个操作也可以用于我们前面的提取启动子fa后,把一个fa合集文件变成多条fa。
$ awk '/^>/ {OUT=substr($0,2) ".fa"}; OUT {print >OUT}' test.fasta
$ ls
AT1G01010.fa AT1G01020.fa test.fasta
脚本出处来源
然后你需要做一个seed文件,我这里抄了BSgenome.TAIR的,只不过把路径变成了我的。
为了大家方便,我这里都是在win10上操作的,所以大家如果自己做应该也没啥问题
Package: BSgenome.Athaliana.TAIR.test
Title: Full genome sequences for Arabidopsis thaliana (TAIR9)
Description: Full genome sequences for Arabidopsis thaliana as provided by TAIR (TAIR9 Genome Release) and stored in Biostrings objects. Note that TAIR10 is an "annotation release" based on the same genome assembly as
TAIR9.
Version: 1.4.2
organism: Arabidopsis thaliana
common_name: Arabidopsis
provider: TAIR
provider_version: TAIR9
release_date: June 9, 2009
release_name: TAIR9 Genome Release
source_url: ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR9_genome_release/
organism_biocview: Arabidopsis_thaliana
BSgenomeObjname: Athaliana
circ_seqs: c("ChrM", "ChrC")
seqnames: paste("Chr", c(1:5, "M", "C"), sep="")
SrcDataFiles
: ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR9_genome_release/TAIR9_chr_all.fas
PkgExamples: genome$Chr1 # same as genome[["Chr1"]]
seqs_srcdir: F:/BS_genome
ondisk_seq_format: rda
注意最后
可能
要留一个空行,
不然会报错
R
In
readLines
(
seed_file
)
:
incomplete
final
line found on
然后开始做
library(BSgenome)
seed_file "F:/BS_genome/BS_genome-seed.txt"
# 看下读的对不对,seed文件
readLines(seed_file)
forgeBSgenomeDataPkg(seed_file)
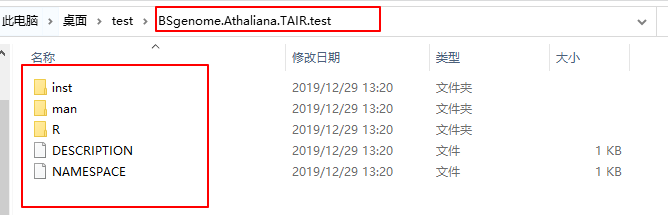
建完之后默认会在你开Rstudio的地方有一个
BSgenome
.
Athaliana
.
TAIR
.
test
文件夹。里面是这样的东西。
然后在终端里面用R CMD,不过R studio也可以用
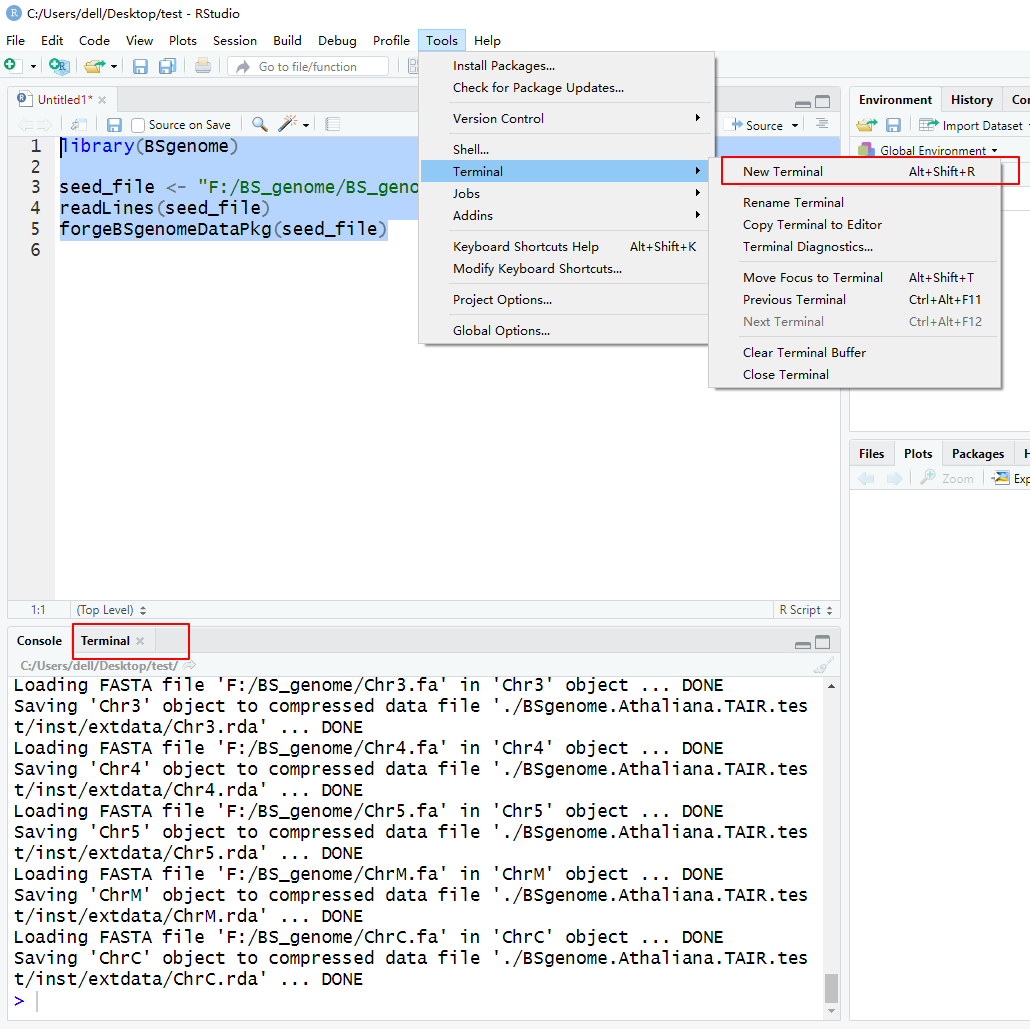
R CMD build BSgenome.Athaliana.TAIR.test
R CMD check BSgenome.Athaliana.TAIR.test_1.4.2.ta
r.gz
R CMD INSTALL BSgenome.Athaliana.TAIR.test_1.4.2.
tar.gz
建完之后就可以正常加载了。然后跟昨天一样了
library(BSgenome.Athaliana.TAIR.test)
seq_length read.table("G:/genome/TAIR10/Athaliana.fa.fai",
stringsAsFactors = F)[,1:2]
Txdb_gtf makeTxDbFromGFF("G:/reference/TAIR10/Araport11_GFF3_genes_transposons.201606.gtf",
chrominfo = Seqinfo(seqnames = seq_length$V1,
seqlengths = seq_length$V2))
gene_gtf genes(Txdb_gtf)
promoter_gtf promoters(gene_gtf,upstream = 4000,downstream = 500)
promoter_gtf trim(promoter_gtf)
promoter_gtf_part promoter_gtf[c("AT1G01010","AT1G01020")]
promoter_gtf_part_seq getSeq(BSgenome.Athaliana.TAIR.test,promoter_gtf_part)
> promoter_gtf_part_seq
A DNAStringSet instance of length 2
width seq names
[1] 4130 CCCTAAACCCTAAACCCTAAACCCTAAACC...AGCTTACCGGAGAATCTGTTGAGGTCAAGG AT1G01010
[2] 4500 ATCCGCAACAATTCACCAATTGAAGAACAA...GCGAGTGAACACAGATGCGTGGGATGTGGT AT1G01020





