WGCNA(Weighted Correlation Network analysis)是一个基于基因表达数据,构建基因共表达网络的方法。WGCNA和差异基因分析(DEG)的差异在于DEG主要分析样本和样本之间的差异,而WGCNA主要分析的是基因和基因之间的关系。WGCNA通过分析基因之间的关联关系,将基因区分为多个模块。而最后通过这些模块和样本表型之间的关联性分析,寻找特定表型的分子特征。
网上例子千千万,但是大部分都是从文档翻译而来,要用起来还是有些费劲,要深入的可以移步这里:
http://www.stat.wisc.edu/~yandell/statgen/ucla/WGCNA/wgcna.html
下面我将根据TCGA乳腺癌基因表达数据以及乳腺癌压型数据,一步一步的使用WGCNA来进行乳腺癌各个亚型共表达模块的挖掘
#############数据准备#############
首先我们需要下载TCGA 的乳腺癌的RNA-seq数据以及临床病理资料,我这里使用我们自己开发的
TCGA简易下载工具
进行下载
首先下载RNA-Seq:
下载之后共得到1215个样本表达数据
进一步下载临床病理资料
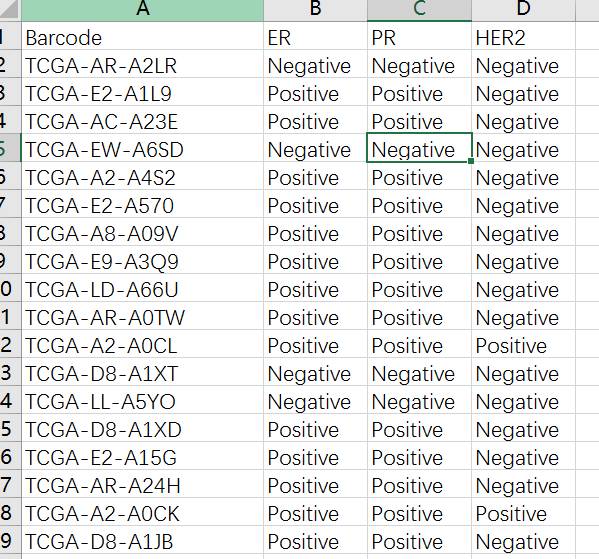
进一步点击 ClinicalFull按钮对病理资料进行提取得到ClinicalFull_matrix.txt文件,使用Excel打开ClinicalFull_matrix.txt文件可以看到共有301列信息,包含了各种用药,随访,预后等等信息,我们这里选择乳腺癌ER、PR、HER2的信息,去除其他用不上的信息,然后选择了其中有明确ER、PR、HER2阳性阴性的样本,随机拿100个做例子吧
 样本筛选完了,现在轮到怎么获取这些样本的RNA-seq数据啦,前面下载了一千多个样本的RNA-seq,从里面找到这一百个样本的表达数据其实也是不需要变成的啦,看清楚咯
样本筛选完了,现在轮到怎么获取这些样本的RNA-seq数据啦,前面下载了一千多个样本的RNA-seq,从里面找到这一百个样本的表达数据其实也是不需要变成的啦,看清楚咯
首先打开RNA-Seq数据目录的fileID.tmp(用Excel打开),然后可以看到两列:
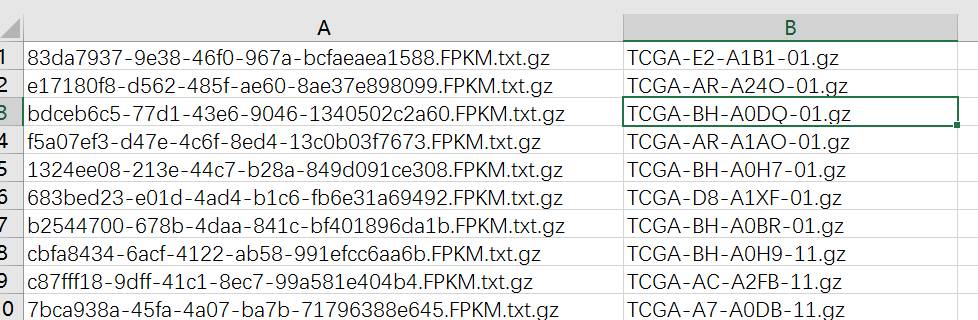
将第二列复制,并且替换-01.gz为空
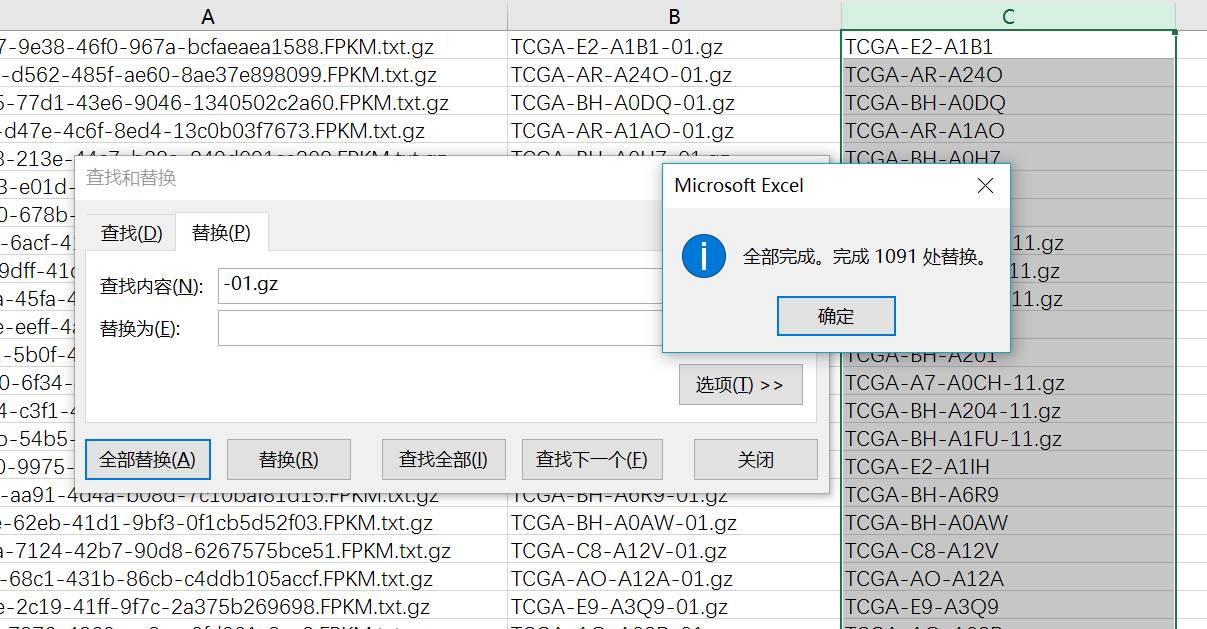
使用Excel的vlookup命令将临床病理资料的那100个样本进行映射
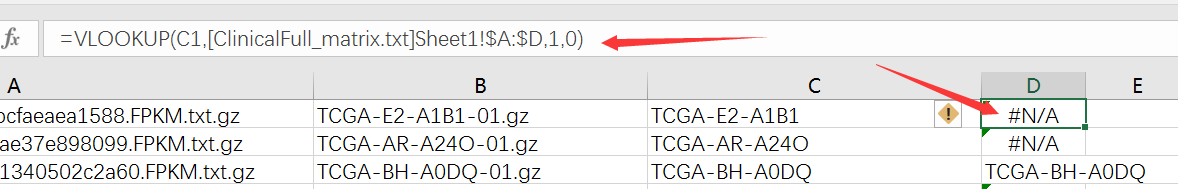
然后筛选非N/A的就得到了这一百个样本对于的RNA-seq数据信息
 进一步删除其他的样本,还原成fileID.tmp格式保存退出:
进一步删除其他的样本,还原成fileID.tmp格式保存退出:
然后使用TCGA简易小工具“合并文件”按钮就得到表达矩阵了,进一步使用ENSD_ID转换按钮就得到了基因表达矩阵和lncRNA表达矩阵了
#################R代码实现WGCNA##############
setwd('E:/rawData/TCGA_DATA/TCGA-BRCA')
samples=read.csv('ClinicalFull_matrix.txt',sep = '\t',row.names = 1)
dim(samples)
#[1] 100 3
expro=read.csv('Merge_matrix.txt.cv.txt',sep = '\t',row.names = 1)
dim(expro)
#[1] 24991 100
数据读取完成,从上述结果可以看出100个样本,有24991个基因,这么多基因全部用来做WGCNA很显然没有必要,我们只要选择一些具有代表性的基因就够了,这里我们采取的方式是选择在100个样本中方差较大的那些基因(意味着在不同样本中变化较大)
继续命令:
m.vars=apply(expro,1,var)
expro.upper=expro[which(m.vars>quantile(m.vars, probs = seq(0, 1, 0.25))[4]),]##选择方差最大的前25%个基因作为后续WGCNA的输入数据集
通过上述步骤拿到了6248个基因的表达谱作为WGCNA的输入数据集,进一步的我们需要看看样本之间的差异情况
datExpr=as.data.frame(t(expro.upper));
gsg = goodSamplesGenes(datExpr, verbose = 3);
gsg$allOK
sampleTree = hclust(dist(datExpr), method = "average")
plot(sampleTree, main = "Sample clustering to detect outliers"
, sub="", xlab="")
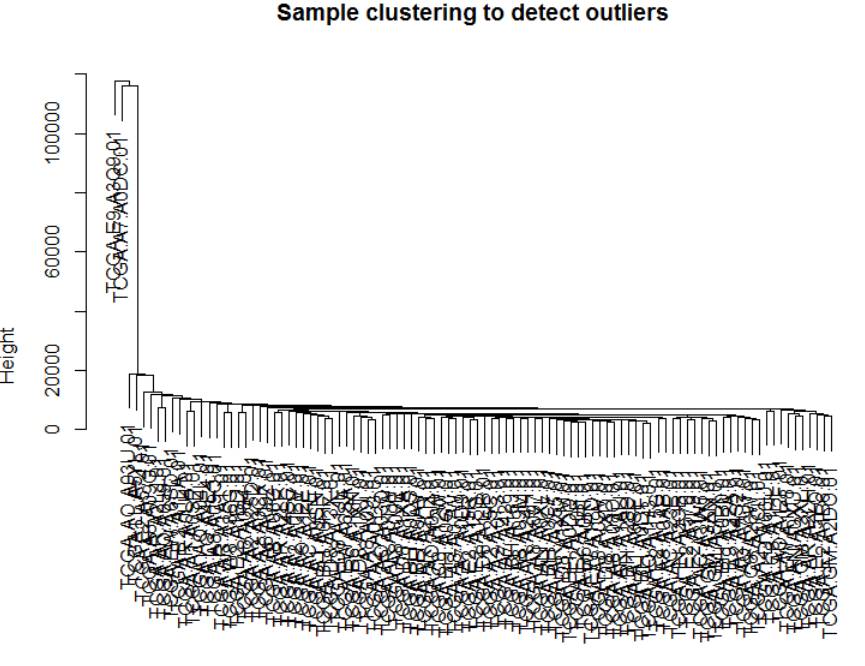
从图中可看出大部分样本表现比较相近,而有两个离群样本,对后续的分析可能造成影响,我们需要将其去掉,共得到98个样本
clust = cutreeStatic(sampleTree, cutHeight = 80000, minSize = 10)
table(clust)
#clust
#0 1
#2 98
keepSamples = (clust==1)
datExpr = datExpr[keepSamples, ]
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
save(datExpr, file = "FPKM-01-dataInput.RData")
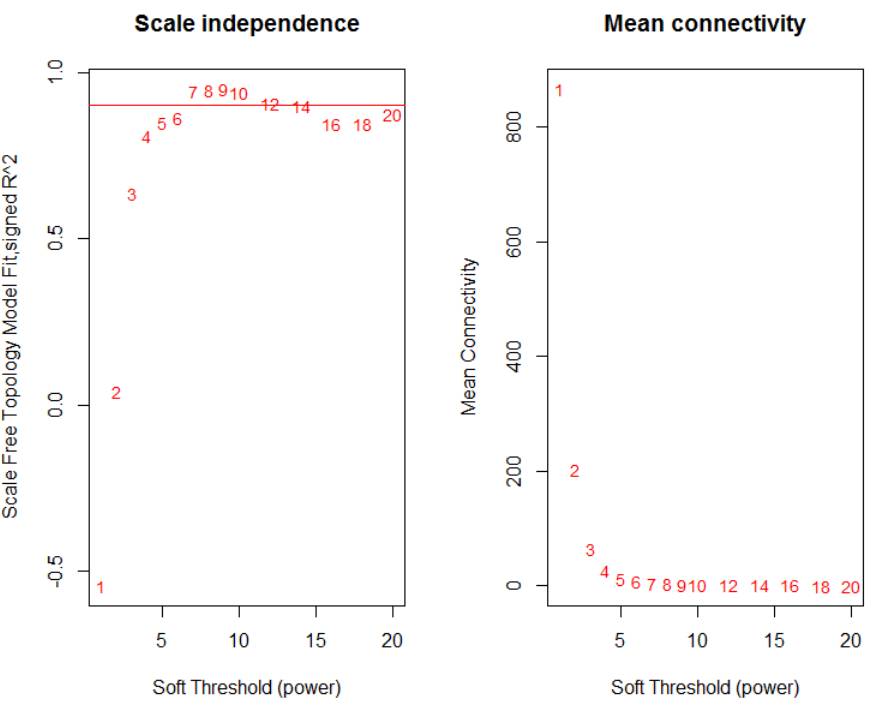
得到最终的数据矩阵之后,我们需要确定软阈值,从代码中可以看出pickSoftThreshold很简单,就两个参数,其他默认即可
powers = c(c(1:10), seq(from = 12, to=20, by=2))
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
##画图##
par(mfrow = c(1,2));
cex1 = 0.9;
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
abline(h=0.90,col="red")
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")