前几
天有个朋友问我,我是怎么理解
2020
版
GCP
对可疑
的非预期的严重不良反应规定的,
我
借此查了一些资料,重新整理
一遍
有关
可疑
的非预期的严重不良反应
的
相关规定和要求,总结
成这
篇小文,与大家分享。
可疑并且非预期的严重不良反应。(来自2020版GCP 第二章
术语 (二十九))
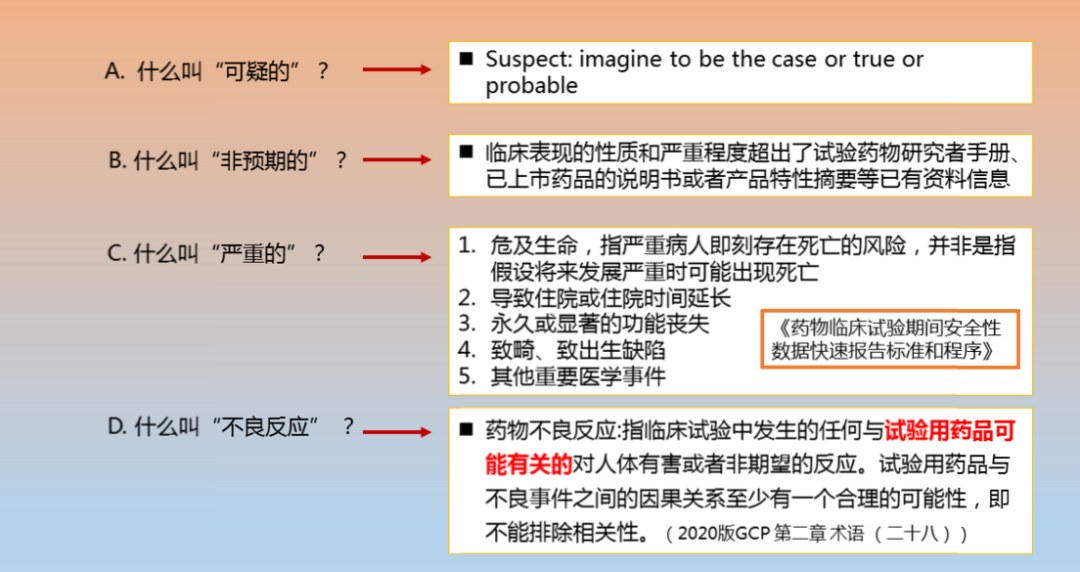 再用逆向思维的方式理解这个定义,什么情况就不是
“可疑
的非预期的严重不良反应
”了呢?
再用逆向思维的方式理解这个定义,什么情况就不是
“可疑
的非预期的严重不良反应
”了呢?
用正向思维和逆向思维两种方式来解释这个定义,不知道你理解起来是否更容易呢?
2020版GCP
对“可疑的非预期的严重不良反应
”要求
的相关章节汇总如下:
为了使报告的流程更清晰,我将上述文字描述的内容,绘画成了下面的报告流程图:
2020版GCP
对“可疑的非预期的严重不良反应
”报告
的时限和报告形式并没有具体的规定。下面是有关上报时限的规定,摘自
ICH E2A
(CDE翻译版)
:
1.
致死或危及生命的非预期不良反应
申办者应在首次获知后尽快报告,不能晚于
7天
,应尽快(通过电话、传真、书面等)通知监管机构,在随后的
8天
内递交信息尽可能完善的随访报告。报告应包括对该发现的重要性及意义的评价,包括有关同类或相似药品的先前经验的资料。
2.
所有其他严重的、非预期的不良反应
死亡和危及生命之外的其他非预期严重不良反应,假如符合快速报告要求,申办者应在首次获知后尽快报告,不能晚于
15天
。
由于报告时限的要求,首次报告时可能来不及收集法规要求的全部信息,出于监管的目的,只要满足以下最低标准,就应该在规定的时限内递交首次报告:摘自
ICH E2A
(CDE翻译版)
4.
不良事件或结局:不良事件可认为是严重的和非预期的,有合理的可疑的因果关系(可以从
“可疑
的非预期的严重不良反应
”的
定义上理解)
除了
以上最低
报告
标准外,其他更多更
详细
的报告信息,可参考:
ICH E2A Attachment 1
:
Key Data Elements for Inclusion in Expedited Reports of Serious Adverse Drug Reactions
.
1.
《药物
临床试验质量管理规范
》
2020
版
2. ICH E2A
《
Clinical Safety Data Management: Definitions And Standards For Expedited Reporting
》
3. ICH E2A
《临床
安全性资料的管理:
加速
报告的定义和标准
》(
CDE
翻译
版
)
4.
《药物临床试验期间安全性数据快速报告标准和程序》
2018
年
5. ICH E2B (R3) Data Elements and Message Specification
点“在看”,送我一朵小黄花






