

Processes in drug discovery and development
众所周知,创新药物的研究关键在于药物的发现。但是,药物的发现则是不可控的过程,具有很大的随机性。而药物筛选是指对可能作为药用的物质进行初步药理活性的检测和试验。相比之下,偶然发现并不可靠,药物筛选才是药物研究中极其重要的部分。随着基因组学、合成化学等领域高通量方法的出现,药物筛选者面临着愈来愈多的新靶标或潜在的有效成分。在医学及其相关学科的发展下,大量分子细胞水平的药物筛选模型不断出现,并应用到药物筛选和研究中,使细胞分子筛选方法逐渐实现一物多筛和多物一筛,再配合先进的检测技术,最后形成了高通量药物筛选技术。
高通量筛选(High throughput screening,HTS)技术是指以分子水平和细胞水平的实验方法为基础,以微板形式作为实验工具载体,以自动化操作系统执行试验过程,以灵敏快速的检测仪器采集实验结果数据,以计算机分析处理实验数据,在同一时间检测数以千万的样品,并以得到的相应数据库支持运转的技术体系,它具有微量、快速、灵敏和准确等特点。简言之就是可以通过一次实验获得大量的信息,并从中找到有价值的信息。
在过去的20多年里,HTS成为化药开发中小分子苗头化合物发现的主力军。很多FDA批准的化学分子类抗肿瘤药物都可追溯至早期的小分子化合物的筛选,如下表所示。对于药物研发历程而言,从项目的早期发现到最终药物的获批上市,大约需要20年的周期。而HTS是一种高效的确定先导化合物的方式,在现代化药开发过程中的重要性不言而喻。


The evolution of Olaparib
最终上市的药物分子与最初小分子筛选过程中获得的苗头化合物也并非完全对应。在很多情况下,成功上市的药物分子往往是在最初HTS获得的苗头化合物基础上进行了数代的优化过程,PARP抑制剂类药物Olaparib就是一个很好的例子。基础研究发现,BRCA1/2失活突变体细胞依赖于PARP进行DNA损伤的修复,这也导致了PARP合成致死效应的发现。Olaparib最初来源于对Maybridge Screening Collection开展的高通量筛选,并于1992年确定了phthalazinones作为苗头化合物。随后在2005年发现了在细胞中有活性的PARP抑制剂,并在2006年获得了良好的代谢稳定性分子,最终在2008年发现了对BRCA1功能缺失细胞具有选择性毒性的化合物。在该药物研发的每一阶段,研究人员都采用临近闪烁分析试验同其他手段一起来追踪抑制剂的活力。而最初高通量筛选所获得的产物至今仍用于不同功能的高内涵筛选过程中,比如阿斯利康从phthalazinone类PARP抑制剂库中发现的苗头化合物,推进到了PARP1/2/4抑制剂AZ0108的发现。

FDA批准的很多药物具有共同的靶点分子,并且是通过不同的筛选策略所获得。以DNA拓扑异构酶为例,FDA批准的抑制剂类药物有靶向于拓扑异构酶Ⅰ(TOP1)的topotecan、irinotecan及靶向于拓扑异构酶Ⅱ(TOP2)的etoposide、dexorubicin及daunorubicin等,另外还有很多该类别药物处于不同的临床试验阶段。
DNA拓扑异构酶对于很多胞内生理过程,如DNA的复制、转录及染色体重构等涉及到的DNA超螺旋的释放过程至关重要。作为其中的一部分,TOP1负责产生单链的DNA缺口,而TOP2负责产生双链的DNA缺口。TOP1的抑制剂可以捕获处于切割过程中的拓扑异构酶-DNA复合体,并诱发DNA损伤,细胞周期阻滞以及细胞的死亡。第一种拓扑异构酶抑制剂camptothecin来源于美国国家癌症研究所(NCI,National Cancer Institute)在上世纪50年代后期发起的天然产物筛选项目,并在70年代推进到了临床研究阶段,最终因为耐受性因素而终止。与此同时,在二代抑制剂类药物topotecan和irinotecan中,camptothecin的稳定性和溶解性都得到了很大提升。1985年,科学家们最终确定了TOP1是camptothecin的直接作用靶点,可结合于酶-DNA的复合体上;结构生物学的进展使得新一代理性设计的相互作用面类抑制剂分子开发成为可能。
另一个例子为已提交上市申请的TOP2抑制剂vosaroxin(又名SNS-595,voreloxin),该药物来源于机理基础上的表型筛选。喹诺酮是一类抗生素,其作用机理为通过扰乱细菌DNA解旋酶及拓扑异构酶Ⅳ的活力而引发DNA损伤。研究人员对包含有喹诺酮环状结构的抗生素进行了细胞杀伤活性的筛选,所获得的苗头化合物采用小鼠P388细胞系进行了进一步优化,最终得到了vosaroxin分子。随后的机制研究表明,该分子直接插入DNA双链并封闭TOP2的活力,从而导致DND双链断裂的产生。令人感兴趣的一点是,接近30年的靶向拓扑异构酶的药物研发史,基本来自于细胞基础上的表型筛选。

The three principles of target class discovery
对于任何一种新型抗肿瘤药物的开发过程而言,最关键的一环节是要确保靶点是准确无误的。简言之,通过对药物靶点或通路的直接调节看是否能诱发所期望的表型,来判断靶点是否确证。在药物研发投入巨额资金之前,对靶点的确认可以增加成功概率,规避相应的风险。对于某一假定靶点的验证可以通过多种策略实现,如基因敲低或敲除(RNAi,CRISPR/Cas)、靶点蛋白的过表达(gain of function)、同时采用细胞株及小鼠模型(包括肿瘤异源移植模型)等。
高质量的化学探针不仅可以完成靶点确认,而且能够展示分子与靶点的相互作用细节。靶点确证本身并不能保证小分子药物开发的成功,毕竟不同类型的靶点所面临的挑战和困难各不相同,即所谓的靶点“成药性”。不管是生物药还是小分子化药,研发过程中的失败原因都具有多样性;但却是有很多案例摆在对于靶点缺乏认知上。对于药物研发项目而言,启动高通量筛选过程并不是最困难的,更为重要的是需要表明靶点与癌症之间的关联性。

BCR-ABL作为药物靶点及其相应抑制剂imatinib的发现过程是一个有趣的例子。上世纪八十年代,科学研究表明大多数慢性粒细胞白血病病人的粒细胞有两条染色体发生换位,产生了融合激酶BCR-ABL,导致了该激酶活性的持续激活,从而造成细胞失控性增生。在此基础上,美国Oregon健康科学大学的生物学家Druker博士提出了以BCR-ABL作为药物靶点的假想,提出开发该靶点的激酶抑制剂治疗慢性粒细胞白血病。该药物的最初分子骨架并不是作为BCR-ABL抑制剂来进行开发,而是由Ciba Pharmaceuticals作为蛋白激酶C的抑制剂进行开发。进一步的优化,获得了一类PDGFR酪氨酸激酶抑制剂分子,该分子同时具有Abl激酶抑制活性。在此基础上,化合物结构进一步进行修饰优化来提高其溶解性和生物利用度,最终获得了imatinib并进入了临床应用。从辈份上来说,最终成为临床药物的imatinib是最初HTS所获得苗头化合物的孙辈,并且靶向于不同的激酶分子。
高通量筛选中所用到的药物发现策略通常可分为细胞基础上的表型筛选及靶点基础上的生化筛选。下文将对上述不同策略及相应案例进行简要概述,并将论述重点放在新型检测手段上,以及两者如何融合来推进药物研发进程。在文献中,一个普遍的共识是基于细胞的表型分析与表型变化的机制是不相关的。与之相反,基于靶点的药物发现往往采用生化或细胞基础上的检测,直接作用于已确认的靶点分子。回顾现有的看肿瘤药物发现历程,基于靶点以及基于表型的高通量筛选方法都扮演了十分重要的作用。
A 基于靶点的筛选(Target based screening)

Target-based approaches for discovery of biologically active small molecules
在绝大多数情况下,采用纯化后的蛋白开展高通量筛选检测具有很多优势。当某一蛋白分子的酶活性或与其他分子间相互作用已被清晰阐明后,通过纯化后的重组蛋白就可以来筛选能够影响某项蛋白功能的化合物分子。筛选过程可以采用不同的生化检测形式:如采用荧光标记产品可以直接读取结果;同报告基因偶联来间接检测;或偶联到单独的光谱、质谱设备等来直接定量反应产物。采用生化检测方法的另一个优势是可以有偏好性的发现一些具有独特机理的抑制剂。对于一个具有辅因子,如NADPH的酶分子而言,我们通常希望获得结合在辅因子结合位点之外的抑制剂;这时可以通过增大辅因子浓度,使其远超KM浓度的情况下开展筛选试验。

蛋白与蛋白间相互作用(PPIs,protein-protein interactions)是公认的比较难于开发药物的靶点,但在药物开发中具有很重要的地位,特别是需要破坏蛋白复合体的形成或信号转导通路的药物领域。Venetoclax由艾伯维公司研发,于2016年4月11日获美国FDA批准上市,又于2016年12月5日获得欧洲EMA批准上市,商品名为Venclexta®,用于治疗带有17p基因缺失突变并且曾接受过至少一种治疗的慢性淋巴细胞白血病患者。Venetoclax选择性的结合BCL-2分子而阻止其同促凋亡蛋白的结合,从而诱导CLL肿瘤细胞的程序性死亡。对于PPI及其他分子间的相互作用可以采用邻近分子信号传递相关的检测方法,这些手段的共性是信号对于标记分子的结合或解离十分敏感,常见的有荧光共振能量转移(FRET,Förster resonance energy transfer),时间分辨荧光能量转移(time-resolved fluorescence energy transfer),放大化学发光亲和均相检测(ALPHA,Amplified Luminescent Proximity Homogeneous Assay)及荧光偏振(fluorescence polarization)。很多抗肿瘤药物作为相互作用面抑制剂,通过与靶点间形成三元复合体,稳定毒性中间体的状态而破坏蛋白-蛋白间相互作用(微管蛋白抑制剂类药物)或蛋白-DNA间的相互作用(拓扑异构酶抑制剂类药物)。用以筛选相互作用面抑制剂的检测方法必须对大分子复合体的稳定性十分敏感,而这一点在目前的高通量筛选及药物发现过程中应用尚不多见。采用体外纯化的靶点蛋白开展研究还可以采用诸如热力学和动力学,以及结构生物学研究等生物物理手段来表征先导化合物特征。
B 机制明确的表型药物发现(MIPDD:Mechanism-InformedPhenotypic Drug Discovery)

MIPDD是基于靶点的小分子筛选路线的一种变通形式,整个实验过程基于细胞模型,但预期的靶点作用机制比较明确,是介于表型筛选和基于靶点的药物发现之间的一种药物发现模式。很多采用基因工程改造细胞系的检测方法可用于MIPDD,包括报告基因分析、高内涵检测、蛋白互补实验等。在很多案例中,MIPDD平台用于二代药物的理性设计改造,如来源于doxorubicin的epirubicin分子。另一个例子为抗雌激素类药物fulvestrant,该药物的筛选模型为能够结合雌激素受体而不激活正常的激素响应的转录活性。筛选过程采用同位素标记的雌激素受体结合实验完成,目标化合物需要不能引发下游的雌激素效应;采用常规的生化检测方法或纯正的表型筛选较难实现这一过程。
另有一点值得重视,MIPDD方法也存在一定的缺陷性,即诱导产生某种期望表型的化合物,未必真正作用在目标信号通路分子上。对于筛选过程而言,重要的是如何能够真正表征对于靶点的作用过程。目前科研人员也正致力于开发各种新型检测策略,以克服上述缺陷。另外,尽管该技术还需进一步的改进来提高筛选通量,改善自身固有的缺陷;但该技术具有自身独特的优势,能够直接筛选药物对胞内靶点分子的作用情况,因而有望使一些不便于获得的靶点变得具有“成药性”,因而潜力巨大。
C 基于表型的筛选(Phenotypic-Based Screening)

Phenotypic-based approaches for discovery of biologically active small molecules
基于表型的筛选方法可以确定能够产生某一特定表型的不同活性化合物,这些分子可能具有各自不同的作用机理;这一方法也是目前肿瘤药物发现领域的中流砥柱。值得关注的是,基于细胞实验的活性检测会表明活性化合物同靶点间的相互作用;当该靶点位于胞内时,该方法的优势会更加明显,其中之一便是该检测结果更容易转化至动物模型乃至最终进入临床药物应用。
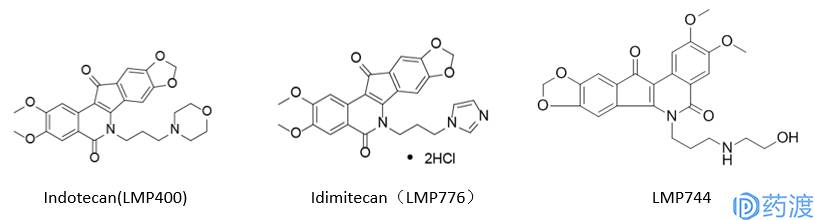
在过去的50年里,药物研发领域最常用于筛选的表型为差异化的细胞死亡或生长停滞。这其中最典型的就是NCI所提供的NCI60细胞库,在同一实验中评估生长相关的三个参数:生长抑制;总体生长抑制和致死浓度。NCI-60为公共可获取的药物研发资源,并已产生了很多重要的科研成果。例如,对1429种抗肿瘤候选药物的全面研究揭示了转运蛋白在肿瘤耐药性中所扮演的重要角色;另外对药物活性及作用规律的比较研究也推动了拓扑异构酶Ⅰ抑制剂LMP400 (indotecan),LMP776 (idimitecan)和LMP744的发现。目前网上有免费资源库CellMiner (http://discover.nci.nih.gov/cellminer),里面集成了NCI-60细胞库所对应的药物活力、基因表达情况及miRNA表达数据,可用于辅助发现“me-too”类化合物。
2016年,在被全球研究人员大量使用了25年后,美国国家癌症研究所(NCI)决定让NCI-60从其药物筛选程序中“退休”。自1990年起,业界和学术界利用NCI-60筛选出10万余种化合物,以研究癌症的分子细节并找到治疗癌症的药物。然而,正如很多其他癌细胞系,NCI-60已在培养基中生存了上千代。这种环境同细胞系的原生环境相差很大。随着时间的流逝,这些细胞适应了塑料培养皿的环境,并且改变了其基因构成和行为。NCI将继续向研究人员提供NCI-60细胞系,但最终将把药物筛选重新聚焦于更新的模型,“来自病人的异种移植”(PDX)模型。
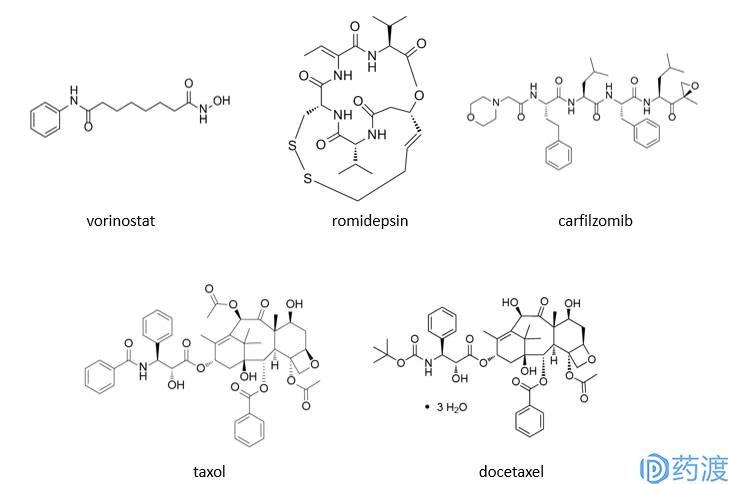
采用单纯的表型筛选(靶点信息未知情况下)最终走向药物发现的案例还有很多,包括组蛋白去乙酰化酶vorinostat、romidepsin及蛋白酶体抑制剂carfilzomib等。Romidepsin最初分离鉴定自
Chromobacterium violaceum
的天然提取物,能够逆转ras转化细胞的形态。随后研究表明,该化合物可以诱导猴病毒40启动子驱动的氯霉素乙酰转移酶报告基因;并具有较强的细胞毒性,能够诱发细胞周期阻滞。在获得美国FDA批准上市之前,该药物由美国国家癌症研究所研发。与此同时,机制研究发现细胞内组蛋白乙酰化水平明显上调,表明该化合物的作用机制为抑制组蛋白去乙酰化酶。如上所述,尽管表型筛选药物发现过程中具体作用机制未知,但作用机制的阐明可以为将来基于靶点的药物发现奠定基础。紫杉醇是该类型药物研发中的一个很好案例,微管蛋白作为紫杉醇作用靶点的确认,促进了二代药物多西紫杉醇及其他一些候选药物的发现。
D 联合用药筛选(Combination Screening)
联合用药在肿瘤的化疗治疗方案中十分普遍,不同的药物同时或先后次序的用于患者治疗。通常的临床试验设计思路为在标准治疗方案中引入新型药物,来评估联合用药的效果。通过筛选发现具有协同效应的药物组合已成为未来药物发现的重要趋势。目前最直接的策略称之为“curve-shift assay”,具体为将细胞用亚毒性剂量的待试药物处理,同时筛选化合物库中的分子;假如肿瘤细胞对所接触的两类药物分子组合比较敏感,就可以通过某种特征筛选出来,该组合可作为进一步研究的对象。这种策略对于筛选目前获批上市的药物库潜力极大,有望加速药物从临床到获批上市的周期。

Curve-shift assay也存在一定的局限性,即单个剂量的受试药物未必是最佳浓度能够被检测到存在协同或抑制效应。简单考虑,可以采用多个浓度的药物来进行测试,但这种多维度的组合测试需要大量的微孔板开展实验。有些科研人员采用如下策略部分解决了该问题:
1)
建立机制明确的集中于抗肿瘤领域的药物库;
2)
采用多孔板将整个流程用机器实现自动化。在一项采用包含有495种化合物库同BTK抑制剂ibrutinib联合用药治疗弥漫型大B细胞淋巴瘤的筛选中,研究人员发现PI3K抑制剂BKM-120同ibrutinib之间存在很强的协同效应,目前该药物组合已进入临床研究。与此同时,其他一些目前已用于临床的药物,如etoposide,doxorubicin及dexamethasone等也表现出较好的协同效应。而在后续采用同一种筛选体系的筛选中,研究人员发现BET蛋白的抑制剂同ibrutinib存在较好的协同效应,并且该协同效应已在肿瘤异源移植模型中得到了进一步证实。采用机制明确的药物库优势在于,较为容易确定联合用药的作用靶点及背后的生物学机制。
2014年1月份,美国FDA批准了来自GSK的2种药物Tafinlar和Mekinist,这2种靶向药物作为联合用药的一个案例,应用于黑色素瘤的治疗。Tafinlar和Mekinist之前被单独批准用于治疗编码B-Raf酶的基因BRAF发生突变的病人:其中Tafinlar及Mekinist各自在不同的生物途径中起作用,Tafinlar为BRAF抑制剂,适用于携带BRAFV600E突变的患者的治疗;Mekinist为首个MEK抑制剂,适用于携带BRAFV600E或V600K突变患者的治疗。在临床试验中,2种药物联合用药的疗效强于Tafinar单独用药的疗效。
2015年春,美国国家癌症研究所规划将建立一个有关联合用药的数据库,被命名为NCI年鉴。这个数据库在一定程度上可以帮助研究者找出哪些药物可能在与别的药物联合应用时比单独用药具有更好的疗效。数据库源自NCI开始于数年前的一个雄心勃勃的项目,项目旨在推动联合用药方法的合理化,NCI就美国FDA批准的每一个抗肿瘤药物组合,进行抗癌细胞系测试,从而建立一个矩阵,绘制出每个药物组合的疗效。这些数据作为一个网络工具可以被药物研究者利用,以分析哪一个药物组合对细胞系的抵抗能力强以及哪一个组合可能是不利的。
E 患者来源的原代细胞用于个性化治疗及药物发现
多项研究已经证实,实验室中传代保存的肿瘤细胞系同它们最初来源的肿瘤细胞已经产生了较多的性质差异;造成该变化的原因很多,包括培养方式不同、基因变异、来源肿瘤的误诊、培养过程的交叉污染以及缺乏体内生理环境的复杂性所带来的改变等。近期有文献报道,目前最为常用的卵巢癌细胞系已经在多项指标上同最初的高级别恶性卵巢癌基本上没有任何相似性。更为糟糕的是,所有六种用于肿瘤药物研究的腺样囊性癌细胞株都受到其他常见细胞株的污染,细思恐极。这些问题都表明了采用患者来源的原代细胞开展筛选的重要性,无论从临床相关的角度还是患者个性化治疗方案的角度。
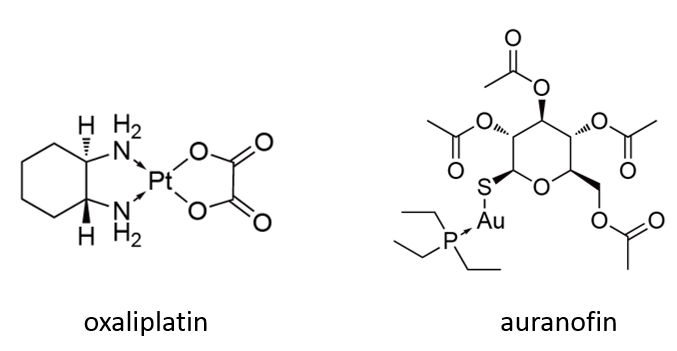
任何恶性肿瘤原代细胞的培养都面临独特的挑战,但目前血液及实体瘤已开始应用于药物研发。在一项采用174名腹膜癌患者样本细胞进行的抗肿瘤药物筛选中,研究人员发现oxaliplatin活性表现出色,经过随后的不同研发阶段,目前该药物早已投入临床应用。而另一项采用原代细胞进行的表型筛选中,对CLL患者样本表现良好的苗头化合物最终导致了auranofin抗肿瘤活性的发现,目前正处于二期临床研究阶段。

Distribution of cancer types in the PDX of Novartis Institutes for Biomedical Research
保持肿瘤细胞原有特性的另外一种策略是构建肿瘤细胞异源移植模型(PDXs,patient-derived tumor xenografts),即将患者肿瘤的原代细胞直接移植到免疫缺陷的小鼠体内,并连续在动物与动物间转移保存。这在科学史上有点像是一种复古的策略,因为早在组织培养筛选技术出现之前,最初用于确定药品活性即采用移植同系啮齿类动物的肿瘤,很多FDA批准上市的药物即采用此类模型所开发。近期Novartis报道采用PDXs模型进行了大量的筛选工作,如上图所示。该策略的优势在于可以将药物的敏感性同基因信息相关联,从而有助于确定耐药性的作用机制,以及发现新的药物作用靶点;局限性在于暂时不便于开展高通量筛选。另外,Novartis所筛选的化合物大部分为药物化学产品,其药物代谢及药代动力学等参数较为明确,因而更接近于机制基础上的药物筛选,而非表型筛选。
F 不同的肿瘤筛选模型
肿瘤多细胞球体示意图
常规二维的组织培养方式并不能反应生理条件下肿瘤的生存环境,因此多细胞肿瘤球体模型应运而生;该模型通过在低附着性环境下培养肿瘤细胞而成。多细胞肿瘤球体可以全面反映肿瘤组织异质性的特征,如核心位置的缺氧,高间质压以及代谢产物积累所引起的酸化过程;同时还可模拟很多药物对肿瘤组织有限的渗透性。另外在某些应用中,还可以采用不同的细胞来制备肿瘤细胞球体,如采用成纤维细胞作为支撑细胞,或利用患者样本及PDXs样本作为球体的核心,能在临床前研究中发挥更大的作用。
采用肿瘤细胞球体作为筛选平台同样面临多项挑战,包括如何在HTS用多孔板中不断制备出具有较为一致生理及生物活性的球体;而球体的物理直径往往会限制他们在96孔及384孔板中的应用。已有的筛选试验表明,某些药物对三维的肿瘤细胞球体及单层培养细胞具有不同的杀伤力;还有些药物对富含肿瘤干细胞的球体具有选择杀伤性。另外,高内涵筛选技术的进步,使得同一筛选过程可以观察多项参数,诸如球体的直径、活/死细胞数统计、细胞凋亡的诱发状态以及直接通过荧光观察药物的渗透情况等。
细胞的迁移及侵袭对于检测方法构建而言是一个不小的挑战。近期科研人员制备了一种多层次的细胞培养物来模拟卵巢癌转移的微环境,包含有原代人间皮细胞、成纤维细胞和胞外基质,用于384孔及1536孔板的筛选。荧光标记的卵巢癌细胞系加入到该筛选体系中,并用高内涵图像采集系统对肿瘤细胞的侵袭进行直接的观察。该工作同时也反映了在HTS筛选平台中引入最新肿瘤筛选模型的巨大潜力。

类器官示意图
类器官(Organoids)模型是一种3D细胞培养系统,其与体内的来源组织或器官高度相似。这些3D系统可复制出已分化组织的复杂空间形态,并能够表现出细胞与细胞、以及细胞与基质之间的相互作用。理想状态下,类器官与体内分化的组织具有相似的生理反应。这不同于传统的2D细胞培养模型,后者在物理、分子和生理学等特性上通常与来源组织的相似性很低。这些模型的目的是为了从分子水平到细胞、组织、器官或整个有机体来阐述身体的功能。目前已成功建立了人与鼠胃、小肠、肝脏、胰腺、前列腺等类器官模型。这些类器官模型均能进行长期培养,且具有稳定的表型和遗传学特征。类器官的培养为我们建立了更可靠的疾病模型,而且类器官也可作为筛选药物的新途径。类器官的培养不仅开发了一种检测细胞介导的和药物引起的疾病模型新方法,同时也开辟了基因和干细胞治疗疾病的新途径。
缩略词表:
AM:acute myeloid leukemia
CLL:chronic lymphocytic leukemia
DTP:Developmental Therapeutics Program
FDA:Food and Drug Administration
HTS:high-throughput screening
IDH:isocitrate dehydrogenase
MIPDD:mechanism-informed phenotypic drug discovery
NCI:National Cancer Institute
PARP:poly(ADP-ribose) polymerase
PDX:patient-derived tumor xenograft
PPI:protein–protein interaction
参考文献:
1. Targetdeconvolution of bioactive small molecules: the heart of chemical biology anddrug discovery. Arch Pharm Res. 2015 Sep;38(9):1627-41. doi:10.1007/s12272-015-0618-3.
2. DrugDiscovery by Molecular Imaging and Monitoring Therapy Response in Lymphoma. IntJ Mol Sci. 2017 Jul 27;18(8). pii: E1639. doi: 10.3390/ijms18081639.
3. Small-MoleculeScreens: A Gateway to Cancer Therapeutic Agents with Case Studies of Food andDrug Administration-Approved Drugs. Pharmacol Rev. 2017 Oct;69(4):479-496. doi:10.1124/pr.117.013755.
4. Thepromise and peril of chemical probes. Nat Chem Biol. 2015 Aug;11(8):536-41.doi: 10.1038/nchembio.1867.
5. Targetidentification for biologically active small molecules using chemical biologyapproaches. Arch Pharm Res. 2016 Sep;39(9):1193-201. doi: 10.1007/s12272-016-0791-z.
6. Principlesof early drug discovery. Br J Pharmacol. 2011 Mar;162(6):1239-49. doi:10.1111/j.1476-5381.2010.01127.x.
7. Targetclass drug discovery. Nat Chem Biol. 2017 Sep 19;13(10):1053-1056. doi:10.1038/nchembio.2473.
8. Phenotypicscreening in cancer drug discovery — past, present and future. Nature ReviewsDrug Discovery 13, 588–602 (2014) doi:10.1038/nrd4366
9. https://en.wikipedia.org/wiki/Phenotypic_screening
10. High-throughputscreening using patient-derived tumor xenografts to predict clinical trial drugresponse. Nat Med. 2015 Nov;21(11):1318-25. doi: 10.1038/nm.3954.
声明:
本文由药渡头条投稿作者撰写,观点仅代表作者本人,不代表药渡头条立场,欢迎交流补充。联系方式:010-82826195 转8048
如需转载,请务必注明文章作者和来源
投稿详
情请点击
“
9-10月 | 王牌写手获奖名单
”







