CNV
分析的工具有好多,令人眼花缭乱,但是有个突出的问题就是目前来说没有哪一个分析工具取得明显的优势,对于笔者这种抱有想挑一个最好的工具使用的心态人来说,研究
CNV
时感觉莫名的苦恼,只能一个一个的试工具并学习这些分析流程。有段时间被
CNVnator
搞得头大,因为该软件安装异常的麻烦,各种报错、
debug
,所以分析工具是否容易安装也是其是否流行的重要因素。
最近笔者发现
2016
年发表在
PLOS computational biology
(
IF=4.542
)上的
CNVkit
目前引用率达到了
74
次,并被很多高分文章引用,该软件主要用
python
编写,容易安装,结果可视化也做的不错。
2017
年
jimmy
大神也写过一篇
CNVkit
的推文,好东西不嫌多写,这里笔者也写一篇
CNVkit
的推文以表对
CNVkit
开发团队的敬意,同时对自己也是以教促学(注:费曼学习法,这两天也是费曼的生日)。
Github地址:
https://github.com/etal/cnvkit
官方教程地址:
https://cnvkit.readthedocs.io/en/stable/index.html
conda config --add channels defaults
conda config --add channels conda-forge
conda config --add channels bioconda
conda create -n cnvkit cnvkit
source activate cnvkit
通过
find
找到
cnvkit.py
脚本的位置,找到其绝对路径,如
/home/anaconda2/envs/cnvkit/bin/cnvkit.py
,以后软件运行就可以用此路径使用
cnvkit
软件了。按官网的建议在
ucsc
上下载了人类
hg38
的参考基因组。同时在本地准备了几个人的
WGS
数据作为练习数据。
官方代码:
cnvkit.py batch *Tumor.bam --normal *Normal.bam
--targets my_baits.bed --annotate refFlat.txt
--fasta hg19.fasta --access data/access-5kb-mappable.hg19.bed
--output-reference my_reference.cnn --output-dir results/
--diagram –scatter
cnvkit.py为运行的脚本
batch是脚本内的一个整合了很多命令的方法,当然也可以使用cnvkit.py提供的access、coverrage、fix等方法一起来完成和batch同样功能的分析,但是作为懒人还是建议使用batch。
Tumor.bam 和Normal.bam都是相应样本的bam文件,建议用bwa通过ucsc的hg38参考基因组做mapping,并用samtool排序转换成bam格式。这里可以输入多个tumor.bam和normal.bam
--targets 想要分析的区域信息
--annotate refFlat格式的基因注释信息,可以从UCSC上下载
--fasta 参考基因组
--access 需要跟bed文件,可用过cnvkit.py access mm10.fasta -s 10000 -o access-10kb.mm10.bed 生成
--output-reference 输出的reference.cnn可以作为下一批tumor数据分析的输入文件,reference.cnn和输入的normal.bam有关
--output-dir 输出目录名
--diagram –scatter 两个都是顺带绘图的参数
在解析好官方的方法后,就可以在自己的服务器上实践了。
代码如下:
/home/anaconda2/envs/cnvkit/bin/cnvkit.py batch /data1/data-sample/human-WGS/bwa-sam-bam/700_bwa.sam.bam --annotate /home/genome/human-ucsc-hg38/ucsc-human-refflat2.txt --normal /data1/data-sample/human-WGS/bwa-sam-bam/699_bwa.sam.bam --method wgs -f /home/genome/human-ucsc-hg38/hg38.fasta --output-reference my_flat_reference.cnn -d 699vs700
首先使用
cnvkit.py
的绝对路径使用脚本,输入了两个
bam
文件:作为
tumor
的
700
和作为
normal
的
699
,输入了
ucsc
上下载的
hg38
的
refFlat
文件,由于是对整个基因组做分析,所以添加了
--method wgs
参数,输入的参考基因组也是在
ucsc
上下载的
hg38
。
运行后会报错(这种报错是我使用的服务器报错,并不代表通用报错),大意是少了
DNAcopy
这个
R
包,进入
R
后通过
bioconductor
下载安装此包。最近
biocondutor
有个问题是部分地方的网络无法使用,可以去官网下载安装包到本地,使用
R CMD INSTALL xxx.tar.gz
安装即可。
再运行还是报错,发现是
refFlat
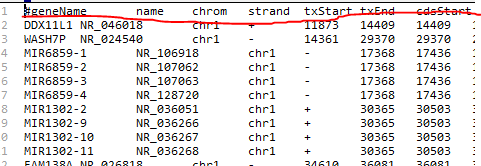
文件有问题,通过查找发现官方提供的范本
refFlat
发现没有第一行的表头,如下图所示,去掉第一行的数据,再次运行就顺利跑完。
运行结果里我们会得到若干文件,其中
700_bwa.sam.cnr
是里面最重要的一个。因为我们参数里没有加包含
--diagram –scatter
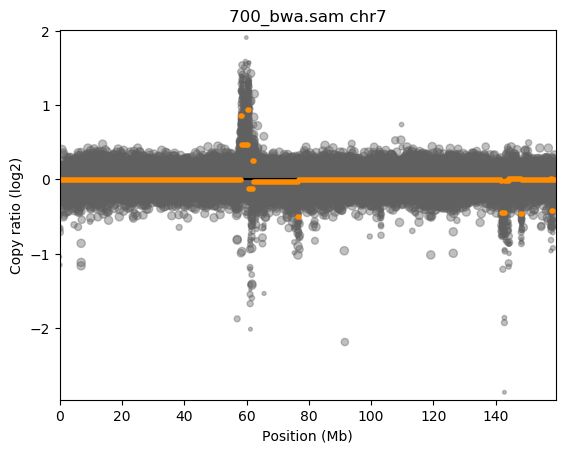
参数,所以默认没有生成可视化图片。我们可以单独用命令行绘制需要的图片,如对
7
号染色体绘制
scatter
图:
绘制scatter图,代码如下:
/home/anaconda2/envs/cnvkit/bin/cnvkit.py scatter -s 700_bwa.sam.cn{s,r} -c chr7 -o scatter-chr7.png
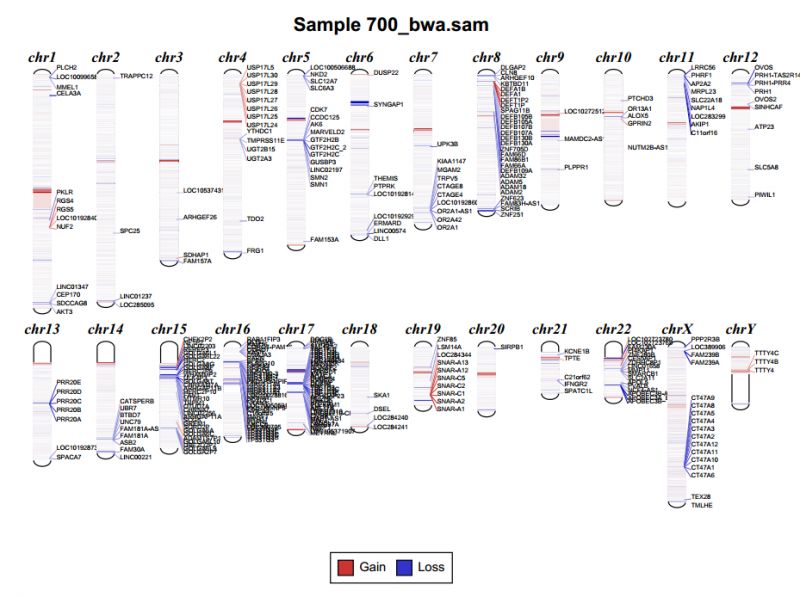
绘制diagram图,代码如下:
/home/anaconda2/envs/cnvkit/bin/cnvkit.py diagram 700_bwa.sam.cnr
结果是一个
pdf
文件,截图如下:
假设我们另一个
tumor
样本
704
要进行同样的分析,则不需要进行第一步的这么多的参数输入,只需要简化一下,使用之前生成的
my_flat_reference.cnn
文件即可,
tumor
样本是
704
,对应的
normal
样本还是
699
,
-p 10
为相应的线程数:
代码如下:
/home/anaconda2/envs/cnvkit/bin/cnvkit.py batch /data1/data-sample/human-WGS/bwa-sam-bam/704_bwa.sam.bam -r my_flat_reference.cnn -d 704 -p 10
从整体上看,笔者输入的
bam
文件平均有
60GB
,但实际运行一个
CNVkit.py
程序的时间约在
1
个小时之内,说明该软件的运行效率还是很高的。
本篇
CNVkit
的使用方法就介绍到这里。
本文为生信草堂原创,欢迎个人转发分享,其它媒体或网站如需转载,请在正文前注明转自生信草堂并联系bioinformatics88
用Atom优雅地写Python
deepin,也许是目前最好的生信练习平台
解决你软件安装疑难杂症的docker





