部分内容节选自:临床研究与医学统计
对于医生来说,如果有某种“特定功能”来预测患者是否会有未知结果,那么许多医疗实践模式或临床决策都会改变。在临床上,几乎每天我们都会听到这样的叹息:“如果我能提前知道,我当然不会这样做!”。举个简单的例子,如果我们可以预测患有恶性肿瘤的患者对某种化疗药物耐药,那么我们将不会选择给患者服用该药物;如果我们可以预测患者在手术过程中可能出现大出血,那么我们将谨慎操作并为患者准备足够的血液制品;如果我们可以预测高脂血症患者不会从某些降脂药物中受益,那么我们可以避免许多无意义的医疗干预。
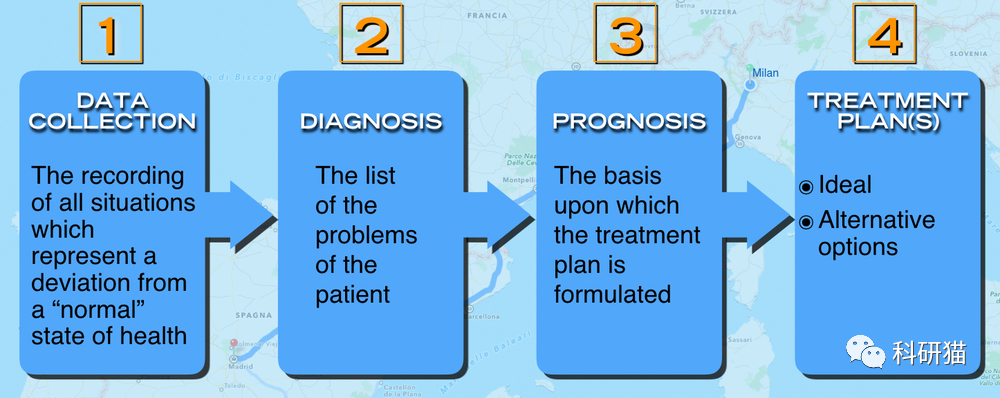
作为一种评估风险和收益的定量工具,临床预测模型可以为医生,患者和卫生管理人员的决策提供更客观,准确的信息,因此其应用变得越来越普遍。在这种刚性需求下,临床预测模型的研究方兴未艾。
随着数据的维度和深度继续变得复杂,对于临床研究的变量选择变得越来越困难。在目前的研究背景和研究趋势下,从临床医生的角度来看,当前的临床预测模型研究分为三种类型。
(1)利用传统的临床特征,病理特征,体格检查结果和实验室检查结果,建立临床预测模型。因为预测变量在临床实践中很容易获得,所以这种类型的模型比其他两种类型的模型更可行。
(2)随着影像组学研究方法的发展,越来越多的研究人员意识到影像的某些表现形式或参数可以代表特定的生物学特征。使用这些大量成像参数,无论是彩色多普勒超声,CT,MR或PET参数,再结合临床特征来构建临床预测模型,通常都可以进一步提高预测模型的准确性。这种类型的模型需要基于筛查影像组学,因此准备的工作量比第一种类型大得多,并且影像组学参数比临床特征更丰富。
(3)随着高通量生物技术(如基因组学和蛋白质组学)的广泛应用,临床研究人员正试图从这些海量生物信息中挖掘生物标志物特征,以构建临床预测模型。这种预测模型是基础医学进入临床医学的良好切入点。比如TCGA和GEO中的大量组学数据。
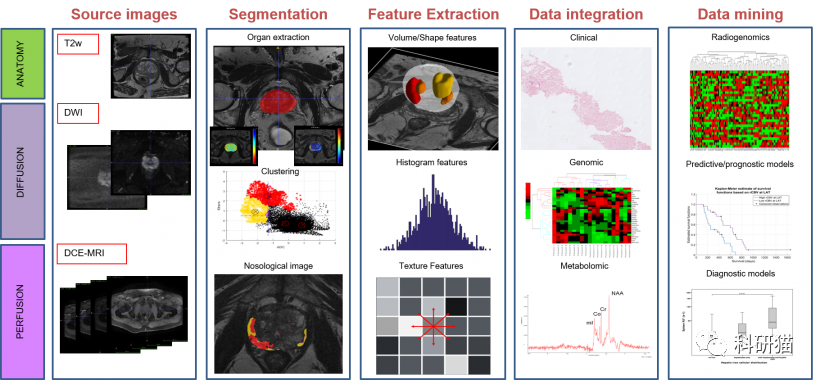
由于第二类和第三类模型中与“组学”相关的特征太多,变量选择非常困难。很难使用传统变量选择方法。那么,有更好的解决方案吗?答案是肯定的。本节中描述的正则化方法是解决方案之一。正则化可以限制回归系数,甚至将其减少到零。现在,有许多算法或算法组合可以用来实现正则化。在这一节中,我们将重点讨LASSO regression。
LASSO 全称是Least absolute shrinkage and selection operator(最小绝对收缩和选择操作)。为了理解Lasso回归,首先我们要理解什么叫做正则化。首先来说,一般的线性模型是Y=β0+β1X1+…+βnXn+e,最佳拟合尝试最小化残差平方和(RSS)。RSS是实际数字与估计数字之差的平方和,可表示为e12+e22+…+en2。我们可以使用正则化在RSS的最小化过程中添加一个新的参数,称为收缩惩罚项(contraction penalty term)。该罚项包含λ和β系数和权重的归一化结果。不同的正则化技术有不同的权重标准化方法。简而言之,我们在模型中用RSS + λ(归一化系数)代替了RSS。我们选择λ,它在建模中被称为调谐参数。如果λ=0,则该模型相当于最小二乘法(OLS),因为所有归一化项都是偏移的。
正则化有什么好处?首先,正则化方法在计算上非常有效。如果我们使用正则化方法,我们只需要为每个λ拟合一个模型,因此效率将大大提高。第二,是偏差/方差权衡问题。在线性模型中,因变量和预测变量之间的关系接近线性,并且最小二乘估计几乎是无偏的,但是可能具有高方差,这意味着训练集中的小变化可能导致最小二乘系数估计结果的大变化。正则化可以适当地选择合适的λ来优化偏差/方差权衡,进而提高回归模型的适应度。最后,系数的正则化也可以用于解决由多重共线性引起的过拟合问题。所谓的正则化,通俗来说就是给平面不可约代数曲线以某种形式的全纯参数表示。这里我简要的画了个参考图。
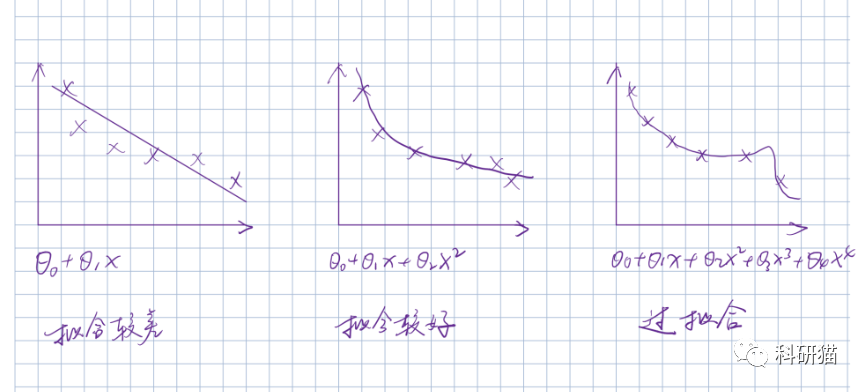
对于同一组数据,我们可以有不同的拟合方法,那么究竟是哪一种拟合方式可以符合我们的实际情况呢?也就是既不会拟合的过于粗糙,也不会拟合的过于精细。把图形转换为公式,就是让θ3和θ4怎么变小,这样就接近我们中间刚刚好的模型了。
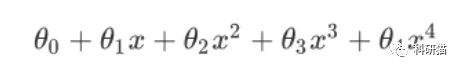
为了解决这个问题,数学家引入了参数lambda,定义模型的惩罚项
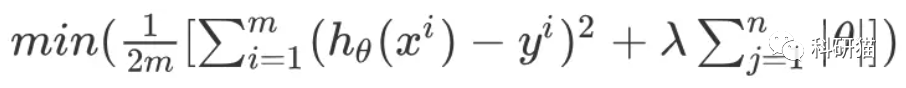
很复杂是不是,简化一下,
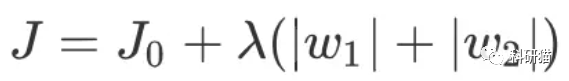
这个方程是不是有点眼熟?高中数学学过|x|+|y|=1,J0的求解过程可以表示为
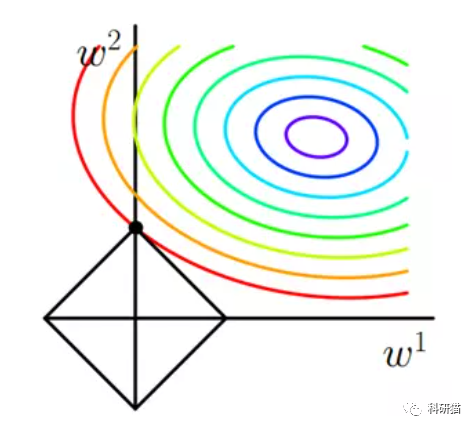
Lasso回归可以使得一些特征的系数变小,甚至还使一些绝对值较小的系数直接变为0,从而增强模型的泛化能力 。对于高纬的特征数据,尤其是线性关系是稀疏的,就采用Lasso回归,或者是要在一堆特征里面找出主要的特征,那么Lasso回归更是首选了。好了,关于基础背景,我们先讲到这里。如果你对上面的“天书”依旧不甚理解,也不要紧,记住一个事情就足够了:什么时候用Lasso?当你的变量太多,难以选择时!
说了这么多,下一步就是大家最关心的部分了,怎么将Lasso回归运用到自己的数据上?下面就带大家来操作一下!首先载入我们的数据:
-
Biopsy Data on Breast Cancer Patients
这是一个MASS包中的乳腺癌活检数据,来自威斯康星医院。这个研究的目的是确定活检结果是良性还是恶性。研究人员使用细针抽吸(FNA)技术收集样本并进行活检以确定诊断(恶性或良性)。我们的任务是开发尽可能精确的预测模型来确定肿瘤的性质。数据集包含699名患者的组织样本,并存储在包含11个变量的数据框中。此数据框包含以下列:
-
ID sample code number (not unique).
-
V1 clump thickness.
-
V2 uniformity of cell size.
-
V3 uniformity of cell shape.
-
V4 marginal adhesion.
-
V5 single epithelial cell size.
-
V6 bare nuclei (16 values are missing).
-
V7 bland chromatin.
-
V8 normal nucleoli.
-
V9 mitoses.
-
class "benign" or "malignant".
好了,了解了我们的实例数据之后,后面就是开始我们的操作啦。第一步呢,永远是读入数据和核查数据。然后,进入到我们此次教程的一个关键环节,分割数据。我们这个数据集当中共有699例样本,我们需要将其随机分割为训练集和测试集,一般的样本比例是2:1,也就是2/3的病人用来做训练,训练出一个准确的模型,剩下1/3病人用来做测试,验证我们的这个模型是否可靠。分割数据的代码如下,整个过程都是随机的:
1
# 分割数据
2
set.seed(
123
)
3
ind 2
, nrow(data), replace =
TRUE
, prob = c(
0.7
,
0.3
))
4
5
# 训练集
6
train 1, ]
#the training data set
7
# 测试集
8
test 2, ]
#the test data set
样本(也就是数据的行)分配好了之后呢,下一步就是把采集到的信息(也就是数据列)进行分配,在我们的这个测试数据中前面V1-V9是采集到的样本信息,最后一列是我们的诊断,也就是肿瘤的良恶性,下面我们把样本信息和样本诊断分割开来:
1
# Convert data to generate input matrices and labels:
2
# x相当于临床信息
3
x 1
:
9
])
4
# y是临床结局
5
y 10]
首先,我们尝试做K-fold交叉验证。在glmnet包中使用cv.glmnet()估计λ值,glmnet默认使用10倍交叉验证。所谓K-fold交叉验证,就是将数据分成k个相同的子集(折叠子集),每次用k-1个子集拟合模型,然后将剩余的子集作为测试集,最后将k个结果合并(一般采用平均值)来确定最终的参数。在此方法中,每个子集仅用作测试集一次。在glmnet包中使用K-折交叉验证非常容易。结果包括每个相应的MSE值和相应的λ。在这里,我们将训练集k值定为5,做5-fold cross validation,这也是比较常用的。
1
# 5-fold交叉验证,找出最佳lambda值
2
fitCV "binomial"
,
3
type.measure =
"class"
,
4
nfolds =
5
)
5
plot(fitCV)
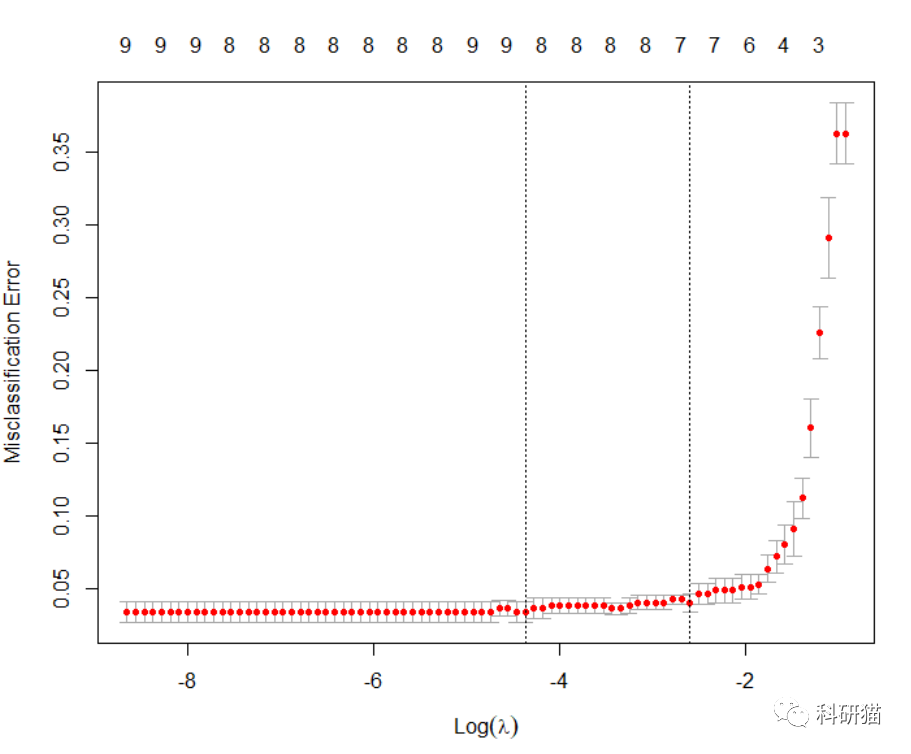
上面的图叫做CV统计图,CV统计图与glmnet中的其他图表有很大的不同,它表示了λ的对数与均方差以及模型中变量数量之间的关系(图49)。图表中的两条垂直虚线表示最小均方误差的对数λ(
左侧虚线
)和最小距离的标准误差的对数λ(
右侧虚线
)。如果存在过拟合问题,那么从最小值到标准误差位置的距离是解决问题的一个很好的起点。
可以看到,我们可以选择的lambda值有两个,具体lambda选值要根据自己实验设计而定。lambda.min是最佳值,lambda.1se则是一倍SE内的更简洁的模型。这也涉及到临床模型一个很重要的考量标准,到底是选择精度高但是复杂的模型,还是放弃部分精度,选择变量更少更有可能有应用价值的模型呢?本例中我们选择最佳lambda值。然后我们来看一下,随着lambda值的变化,每个观察值对应的系数的变化趋势。
1
# check the coef
2
fit "binomial"
, alpha =
1
)
# make sure alpha = 1
3
plot(fit, xvar=
"lambda"
,label =
TRUE
)
4
abline(v = log(lambda.min,
10
), lty =
3
,
5
lwd =
2
,
6
col =
"black"
)
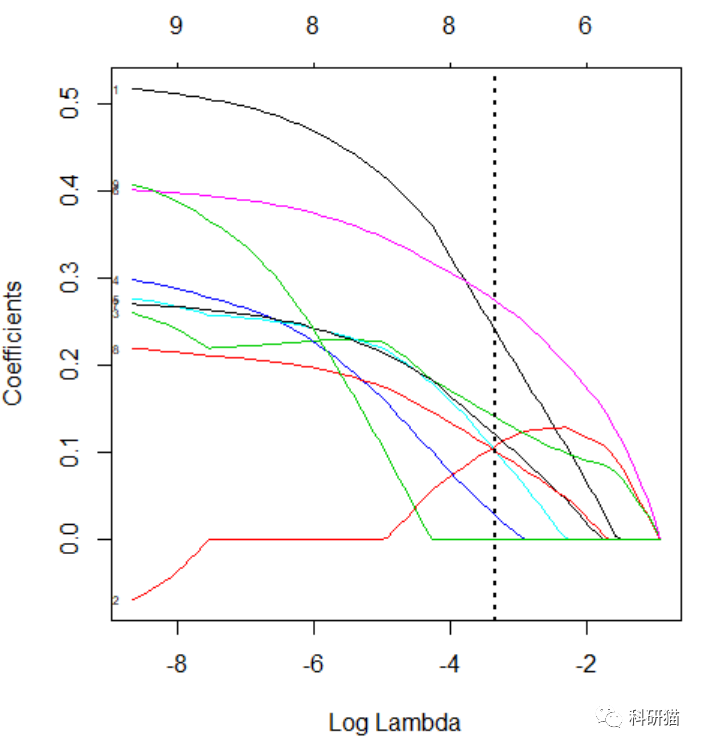
此图显示,随着λ的减少,压缩参数减少,系数的绝对值增加(图44)。这个模型应该如何在文章中描述呢?我们来输出系数:
1
# get the coef
2
coef.min = coef(fitCV, s =
"lambda.min"
)
3
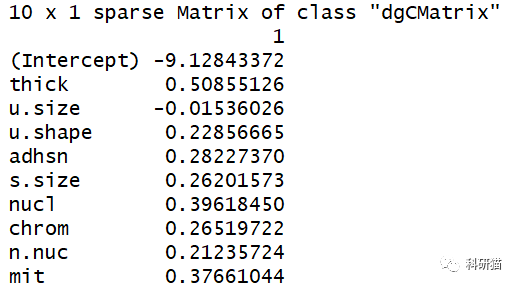
coef.min
然后就会生成每个变量所对应的系数,我们也可以通过这个系数来对模型进行描述。
好了,模型构建好了,下面就是检查这个模型在测试数据中是否能够表现出较好的效能了。我们分别在训练集和测试集中进行测试,然后用ROC曲线来描述模型的统计效能。
1
# 开始进行模型检验
2
#首先是训练集
3
predCV.train 1
:
9
]),
4
s =
"lambda.min"
,
5
type =
"response"
)
6
actuals.train "malignant",
1
,
0
)
7
8
#其次是测试集
9
predCV.test 1:
9
]),
10
s =
"lambda.min"
,
11
type =
"response"
)
12
actuals.test "malignant",
1
,
0
)
通过predict函数就可以实现我们对于模型的预测了,fitCV对象就是我们构建的模型,分别在train和test两个数据集中进行评估。那么评估的结果如何呢?最直接的方法就是看ROC曲线,比较AUC值。
1
## plot ROC
2
x1
3
smooth=
F
,
4
lwd=
2
,
5
ylim=c(
0
,
1
),
6
xlim=c(
1
,
0
),
7
legacy.axes=
T
,
8
main=
""
,
9
col=
"red"
)
10
x2
11
smooth=
F
,
12
add=
T
,
13
lwd=
2
,
14
ylim=c(
0
,
1
),
15
xlim=c(
1
,
0
),
16
legacy.axes=
T
,
17
main=
""
,
18
col=
"seagreen3"
)
19
# check ROC
20
x1[[
"auc"
]]
21
x2[[
"auc"
]]
22
23
# add figure legend
24
legend.name "Train:AUC"
,sprintf(
"%.4f"
,x1[[
"auc"
]])),
25
paste(
"Test:AUC"
,sprintf(
"%.4f"
,x2[[
"auc"
]])))
26
legend(
"bottomright"
,
27
legend=legend.name,
28
lwd =
2
,
29
col = c(
"red"
,
"seagreen3"
),
30
bty=
"n"
)
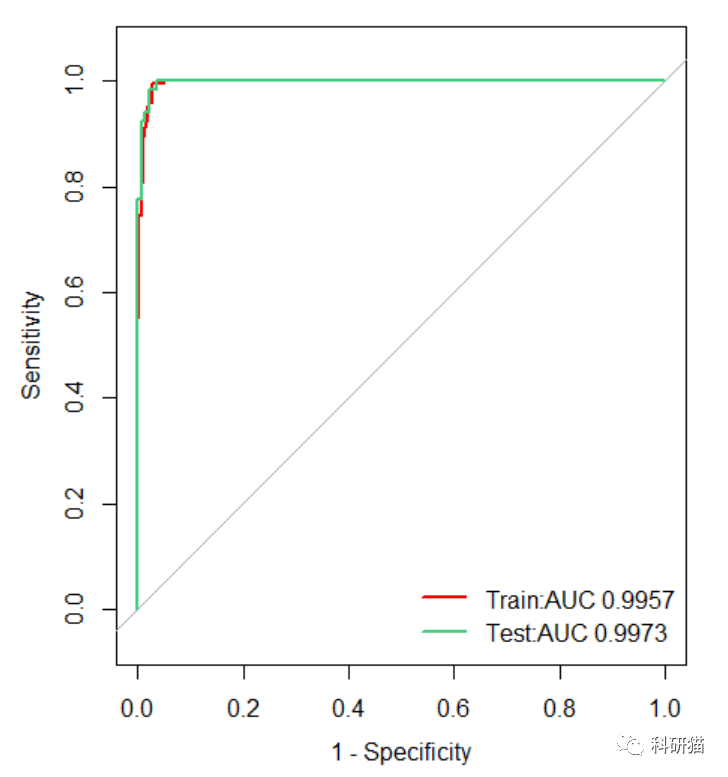
可以看到该模型在训练集和测试集中都表现良好,AUC值都达到了0.99以上。太神奇了,有没有!这么好的诊断模型,哪里去找?感觉有了这个代码,很多文章分分钟呼之欲出啊。
此外,关于ROC曲线的故事,我们已经在另外的系列中跟大家深入讲过了,通过我们科研猫出品的“ROC曲线终结者”代码,不仅可以自动绘制多种组别的模型曲线,还能实现不同模型间的两两比较,详见
【科研猫·统计】ROC曲线(2):一码到底
。今天的LASSO回归就到这里,联系客服领取代码和测试数据哦。
参考文献:Zhi-Rui Zhou, Wei-Wei Wang, Yan Li, et al. In-depth mining of clinical data: the construction of clinical prediction model with R.Annals of Translational Medicine.
本期干货
-
LASSO回归
代码 -
·
关注“
科研猫
”公众号,联系客服领取
往期干货链接
精通R语言(含代码及测试数据)





