
本文转自公众号“肿瘤科医生”,仅供学习交流使用,如有侵权,请联系我们删除,谢谢。
姜时雨 石远凯
中华肿瘤杂志, 2017,39(10) : 721-725.
生物制品是由重组DNA在活体细胞内表达产生的蛋白质,具有分子量大、结构复杂的特点,迄今已有30多年的发展史,主要包括重组蛋白、单克隆抗体、激素和细胞因子等,为人类疾病的预防和治疗做出了突出贡献。现如今生物制药市场份额巨大,2011年,生物制药全球销量估测达1 570亿美元,2016年有望超过2 000亿美元
[1,2]
,而生物制药在全球制药中所占份额将有望在2018年达到49%
[3]
。但是由于研发和制造工艺复杂,研发周期漫长,耗资巨大,导致生物制品价格高昂,根据Silver
[2]
统计,在2012年研发1个新的生物药品可能会耗费至少19亿美元,应运而生的便是生物类似药。
生物类似药是一种与已被批准的原研生物药品高度相似,但不完全相同的生物制品。换言之,如果一个生物类似药被批准,需要证明它与原研药高度相似,在临床上无有意义的差别,也就是在安全性和有效性上与原研药无具有统计意义的差别,临床上无活性部分的轻度差异在允许范围内,主要涉及的生物类似药有干扰素、促红细胞生成素(erythropoietin,EPO)、低分子肝素、重组人粒细胞集落刺激因子(recombinant human granulocyte colony-stimulating factor,rhG-CSF)、胰岛素类似物、人生长激素和单克隆抗体等。同样,研发一个新的生物类似药也耗资较大,耗时7~8年的时间,耗资约1~2.5亿美元,但相对于原研生物制品,已是九牛一毛。随着原研药物专利陆续到期,生物类似药的发展有助于减少医药产业总体支出,推动药品价格良性竞争,提供患者更多的治疗选择,顺应卫生保健事业发展。举例来说,由于EPO的生物类似药研发上市,欧洲21个国家EPO的价格从2006年到2013年下降了35%。由此看来,不久的将来,一场生物类似药的热潮有望席卷全球。
肿瘤领域因其患病人数众多,治疗费用昂贵,治疗发展迅速,受到了广泛的关注,生物类似药生产厂家和各原研药生产商也将抗肿瘤药物作为研发的重点领域。早在10年前,生物类似药在肿瘤支持治疗领域已有一席之地,欧洲联盟自2007年开始,批准EPO和rhG-CSF的生物类似药进入市场,美国也在2015年批准rhG-CSF的生物类似药进入市场,并得到美国国立综合癌症网络(NCCN)指南的认可
[4]
。而今,随着利妥昔单抗、曲妥珠单抗和贝伐珠单抗等生物制药中领军药物的专利先后到期,在肿瘤领域的单抗类生物类似药研发如火如荼,近几年我国对于生物类似药的研发热情,也折射出生物类似药在我国医药行业未来的地位。
1.生物类似药研发现况:
自从2006年第1个重组人生长激素生物类似药(Omnitrope)在欧洲上市,至今生物类似物在全球药品市场已有10年的历史。截至2015年末,已有4大类41种生物类似药被批准上市。世界范围内,生物制药领域日新月异,在过去的10年中,全球共80余种生物药品入市,生物类似药在卫生系统发展的良性循环中起了很大的作用,许多具有竞争力的生物类似药的研发节约了医疗资源,同时进一步激励以改善患者预后为目标的药品创新和研发。
随着生物制药的市值从2002年的460亿美元增长至2020年预估的3 900亿美元,生物类似药也显现出前所未有的重要性
[5,6]
。通过与原研生物药品在越来越多治疗领域中的竞争,生物类似药为药厂、医师和患者提供了更多的机遇和选择,预计在接下来的5年可能会为欧洲5个主要国家和美国累积节约500亿欧元,甚至可能达到1 000亿欧元
[7]
。值得一提的是,在生物制药市场中,2015—2020年专利到期的药物较多,2015年9月销售排行前6位的生物药品将在2015—2020年的5年间失去专利(表1)
[8]
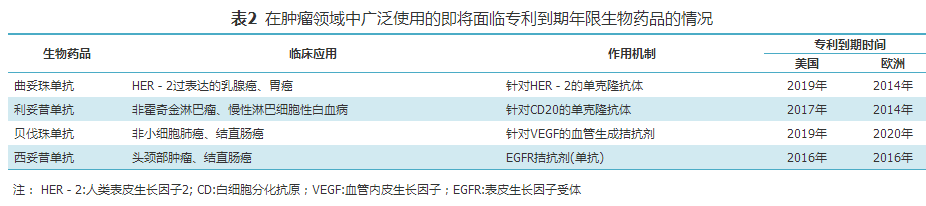
,为生物类似药的研发带来了巨大推动力,其中就有在肿瘤领域得到广泛应用的利妥昔单抗、曲妥珠单抗和贝伐珠单抗(表2)
[9]
。在我国,安全、有效、经济的生物药品是治疗的迫切需求,这就为生物类似药的研发带来了动力。由上海复宏汉霖生物技术有限公司研发的美罗华生物类似药HLX01于2015年通过临床试验审批,目前正在我国开展多中心Ⅲ期临床试验(NCT02787239)。我们可以预见,未来的5年即将见证生物类似药的市场竞争效应,为患者带来效价比更高的治疗选择,让患者能有机会在与疾病抗争的每个节点选择最适合的药物,最终改善患者的预后。

2.生物类似药的评价:
在生物类似药越来越受关注的背景下,鉴于生物类似药的制造者不可能完全复制原研药生产企业的制作工艺,因此,对于生物类似药而言,无法共用小分子仿制药的批准流程,由此诞生了生物类似药审评指南。欧洲药品管理局(European Medicines Agency,EMA)在生物类似药的审评工作上一直走在世界前列,2005年EMA出台生物类似药指南,指南要求不能单纯根据与原研生物制剂可比的生物等效性批准生物类似药,应该提供与原研生物制剂相似的安全性和有效性的非临床和临床研究数据,且证明其与原研生物制剂具有可比性
[10]
。
2010年11月,EMA出台了关于单克隆抗体生物类似药研发指南草案
[11]
,进一步拓宽了生物类似药的研究范围。除了欧洲联盟,美国食品药品监督管理局(Food and Drug Administration,FDA)也于2012年颁布了生物类似药研发指南文件草案,其中涉及到关于诠释生物类似药与原研药相似性的问题,为生物类似药进入美国市场建立了一条快速审批通道。迄今为止,EMA
[11,12]
、FDA指南
[13]
已几经更新,而我国国家食品药品监督管理总局于2015年2月28日发布了《生物类似药研发与评价技术指导原则(试行)》
[14]
,确定了我国生物类似药的监管框架,为生物类似药在中国的研发提供了依据。纵观各国生物类似药研发的历程,对于安全性和有效性的评价贯彻始终,患者的安全性和有效性一直是关注的核心问题,临床前研究和临床研究对于生物类似药的评价需要通过一系列严格、科学的评价体系完成。任何生物类似药进入临床应用的程序中,最核心的环节为证明生物类似药与原研药在结构及功能、药代动力学、药效动力学、安全性和有效性相似的临床前及临床研究(表3)
[12,13,15,16]
。
此外,由于复杂的分子结构和生产过程,生物制品分子结构可能在生产的各个环节中发生改变,导致发生免疫原性、过敏反应和其他不良反应的风险增加。因此,对于生物类似药而言,要求其安全性和原研药类似具有挑战性。简而言之,根据指南要求,生物类似药需要满足以下条件:(1)与原研药具有生物类似性;(2)与原研药已知的作用机制相同;(3)适应证、给药途径和剂量与原研药一样;(4)药品生产企业能保证生物制品的安全性、纯度和有效性。

3.抗肿瘤生物类似药的特殊性:
相对于小分子药物而言,生物类似药具有分子量大、结构复杂等特点,其来源多为活体细胞,对生产环境具有敏感性,细胞培养条件(温度、营养)、产品的加工、纯化、储存和包装中微小的差别均会对产品的质量、纯度、生物特性和临床效果产生影响。生物类似药生产与原研药相似的产品,但无法做到完全一致,而小分子药物与原研药完全一致。对于生物类似药,尤其是单克隆抗体生物类似药,由于蛋白的聚合程度、一级序列的修饰、糖基化和局部二级结构的不同均会影响最终的蛋白产物,即便与原研药的表达载体、技术和生产流程一样,仍然无法保证产品的类似性。生物制品的内在结构、理化性质和生产程序对其安全性和有效性均有潜在的影响(表4)
[9]
。
此外,生物类似药的免疫原性亦关乎药品安全性,全球生物制药企业和药品监管机构均非常关注产品的免疫原性,利用生物技术生产的外源性蛋白来补充体内不足的同种内源性蛋白,可能会激活机体的免疫系统,产生针对外源性蛋白的抗体,而这种抗体可能与人体内源性蛋白产生交叉反应,从而产生不良作用。在某些情况下,这些由生物制药产品引起的免疫反应产生的抗体不引起体内内源性蛋白的交叉反应,但可能会降低药物的有效性,同时可能导致人体产生炎症反应、过敏和血清病
[16]
。例如,人体内源性EPO被抗体中和而失去活性,可能导致单纯红细胞再生障碍性贫血(pure red cell aplasia,PRCA)
[17]
。1998—2003年,许多肾性贫血的患者就是因为使用宜保利血(Eprex)而导致PRCA,该事件提醒人们应该高度重视生物药品免疫原性的不可预测性和长期、广泛使用带来的后果。
由于生物类似药的特殊性,其市场准入的要求将会介于传统小分子仿制药和新的生物药之间,临床研究也面临着诸多的挑战。以抗肿瘤药物为例,临床试验患者选择的难易程度、试验周期以及试验费用等都是必须考虑的问题,生物类似药的等效性研究需要与原研药物对照进行,而参比的原研药物往往价格高昂。此外,还需要考虑伦理问题,例如利妥昔单抗生物类似药Ⅰ期临床试验如果在健康受试者中进行,则不符合伦理原则;早期乳腺癌患者采用生物类似药进行新辅助化疗在伦理学方面仍存在争论。如何通过最少的病例获得尽可能多的信息也是临床研究者与制药企业探寻和努力的方向,生物类似药仅需Ⅰ期及Ⅲ期临床试验,并且Ⅲ期临床试验推荐以总缓解率(overall response rate,ORR)作为首要终点,但ORR是否能充分反映药物的疗效和安全性,以及作为适应证外推的强有力证据,仍存在不同的观点。










