阿里云天池十月之星
作者:
刘钰舒
哈尔滨工业大学研究生、Top级竞赛选手

大牛来历
大家好呀,我叫刘钰舒,天池ID是liuyushu2333。我是一名自哈尔滨工业大学的计算机技术研究生,主攻计算机视觉算法,目前研二。说起来我本科是读应用化学专业的,保送到哈工大之后才开始学习计算机相关的课程。专业转换给我带来了许多挑战,对比其他小伙伴我总是觉得自己在专业技术方面还有很大提升空间。为了弥补这些差距,除了在课堂上好好学习天天向上之外,课余时间里提升自己的一大妙招就是在天池平台参加比赛。我的参赛记录简单如下:
工作+学习分享
从零基础入门的学习赛开始,逐渐到蛋白质结构预测这种略有难度的竞赛,我对知识的运用越来越熟练,经验也越来越丰富,自然的,所获名次也就水涨船高。我喜欢用算法解决实际问题,因为在真实场景下,问题相较课上的练习会变得棘手的多,非常提升我们的知识运用的灵活度和能力。我在天池参加的很多比赛都是真实场景下的,比如饿了么调度算法的比赛,就是不断产生订单,然后你要实时地把订单分配给最合适的骑手。因为是真实场景下的,对相应时间和骑手速度啊背单数量都有很多限制,位置也都是具体的经纬度,写起程序来还挺复杂,然后也经常超时,需要我们对时间复杂度啊做一些优化,非常提升代码的编写能力。此外,对于每个比赛天池官方总是组织很多视频课程啊学习资料啊,比如我当时参加了零基础数据挖掘比赛,通过详细的数据处理、特征工程、建立模型等等视频,我好像突然知道数据挖掘是怎么回事了。论坛也经常有前排大佬分享的各种方案,大家集思广益,真的是能学习到各种厉害的技术。
由于咱哈工大是两年制研究生,我之前就一直在找工作了。目前来看,天池的竞赛经历对就业帮助是非常大滴。首先是投简历时,很多厂的算法岗都会说明简历筛选条件,通常是编程、算法、数据挖掘类比赛获奖优先,我个人也实际感受到我的简历还是比较容易通过筛选的。其次,自我介绍结束以后,面试官通常会让你再深入介绍你参与的一个项目,参加的比赛就可以作为一个项目来说。我是做计算机视觉算法的,投的也都是这个岗位,比如我参加的街景字符识别大赛就经常被问到。感觉计算机视觉算法部门做OCR的比较多,这样会和面试官比较有话说。当然如果有论文或者实习肯定能再锦上添花,但是像我这种没有论文没有实习的同学吧,参加比赛是最效率的选择。因为像论文或者是实习可能需要投入的时间都比较长,而比赛一般只需要十几天左右。此外,比赛结束后无论是否获奖都会有一个很漂亮的名次证书,因为天池的比赛知名度还是挺高的,这个证书算是自己参与项目有力的证明材料。我也遇到过远程面试期间面试官让你发比赛链接,他会详细看看赛题的任务和别的选手的方案,再和你详细讨论这个比赛。最后,面试官也可能会问比赛方案中的算法知识点,像数据挖掘类的比赛一般会深入问一下LightGBM、XGBoost等Boosting模型的原理,我一般是看看用到的模型的源码,看看相关算法的面试题,再结合自己使用模型的感受向面试官介绍,感觉效果会好一点。如果用到了自己搭的深度学习模型,面试官也会问问为什么搭成这样的网络结构而没有加一层或者减一层网络,为什么不用LSTM,为什么没有用更深的比如ResNet。我一般按实际模型选择和调参的实践经验说一说。这些算法知识点都是可以提前准备的,总比人家随便问个碰巧是你不懂的领域好,或者因为你没啥可说的 面试官可能会问一些他自己比较熟悉的领域,那就比较难回答了。所以参加比赛一方面可以把所学理论知识实际运用起来,把知识掌握地更扎实,也可以更有针对性地准备面试,掌握面试的主动性。对于我的话,零论文零实习零项目,基础还不咋好(毕竟学的时间短),真的很感谢天池让我在2021灰飞烟灭的算法岗中和面试官有可以说的东西。
下面我分享下自己在取得蛋白质结构预测大赛top3的名次后写出的经验分享文章,希望能给大家带来帮助。
大赛经验分享
赛题介绍
疫情当前,解析病毒关键蛋白的结构对于开发阻断病毒入侵的药物、找到新冠病毒可能的中间宿主、指导新冠病毒疫苗的研发等有重要意义。天池联合达摩院通过公开蛋白结构数据,让更多跨学科开发者参与蛋白质二级结构预测研究中,为抗击疫情贡献技术力量。疫情当前,解析病毒关键蛋白的结构对于开发阻断病毒入侵的药物、找到新冠病毒可能的中间宿主、指导新冠病毒疫苗的研发等有重要意义。
蛋白质是生命活动中重要的组成,蛋白质的结构决定了蛋白质在生命活动中的功能,因此对蛋白质结构进行分析具有重要的实际意义。蛋白质结构又可分为一级结构,二级结构,三级结构和四级结构。蛋白质特定的空间结构由其一级结构(氨基酸序列)所决定,二级结构为相邻氨基酸形成的折叠旋转等空间结构,三级结构是在二级结构的基础上,进一步形成的特定空间结构,其中二级结构的预测可作为一级结构和三级结构纽带,是从一级结构预测三级结构的关键步骤。此外对二级结构的预测也有助于全新蛋白质的设计,有助于后续实验中对二级结构的指认。目前并非所有发现的蛋白质都有明确的二级结构信息,蛋白质结构主要通过X射线晶体衍射和核磁共振成像技术测定,实验成本较高。随着新的分析技术的发展,利用数据驱动,仅通过蛋白质序列对相应蛋白质结构的预测成为热点,可大大降低实验成本。
数据分析
目前已知一部分氨基酸序列和与其对应的二级结构,通过已有数据寻找一级结构到二级结构的映射模型,提高通过氨基酸序列进行蛋白质二级结构预测的准确性。
这里蛋白质的一级结构为氨基酸序列,由23种字母排列组合而成;二级结构为氨基酸序列的螺旋、折叠、卷曲等等,由7种字母和空格排列组合而成。这里尤其需要注意的是,二级结构和一级结构一样长(包含空格)。训练集一共包含20000条一级结构及其对应二级结构,测试集未知。
特征选择与处理
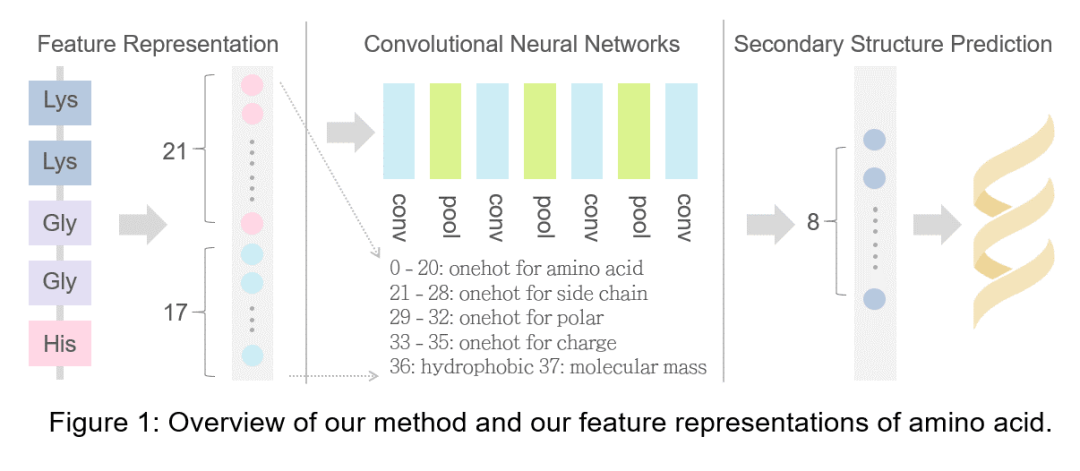 由于自然界中只存在20种常见氨基酸,题目中出现23种,有2种特别少,我们按照生物学对氨基酸的分类标准将其归入X,代表未知氨基酸。对于一个一级结构的字母,我们选择38维的特征向量来表示它,其中前21维为21种氨基酸的onehot,考虑到氨基酸侧链性质对结构影响较大,我们设置后17维为氨基酸的理化性质onehot,这里理化性质主要包括侧链残基的类别,带电性,极性,亲疏水性,相对分子量等,具体数据来源于
Table of standard amino acid abbreviations and properties
,38维中具体每一维特征如下:
由于自然界中只存在20种常见氨基酸,题目中出现23种,有2种特别少,我们按照生物学对氨基酸的分类标准将其归入X,代表未知氨基酸。对于一个一级结构的字母,我们选择38维的特征向量来表示它,其中前21维为21种氨基酸的onehot,考虑到氨基酸侧链性质对结构影响较大,我们设置后17维为氨基酸的理化性质onehot,这里理化性质主要包括侧链残基的类别,带电性,极性,亲疏水性,相对分子量等,具体数据来源于
Table of standard amino acid abbreviations and properties
,38维中具体每一维特征如下:
- 0: G
- 1: P
- 2: T
- 3: E
- 4: S
- 5: K
- 6: C
- 7: L
- 8: M
- 9: V
-10: D
-11: A
-12: R
-13: I
-14: N
-15: H
-16: F
-17: W
-18: Y
-19: Q
-20: X
-21: Side Chain Class is acid
-22: Side Chain Class is aliphatic
-23: Side Chain Class is amide
-24: Side Chain Class is aromatic
-25: Side Chain Class is basic
-26: Side Chain Class is cyclic
-27: Side Chain Class is hydroxyl-containing
-28: Side Chain Class is sulfur-containing
-29: Side Chain Polarity is acidic polar
-30: Side Chain Polarity is nonpolar
-31: Side Chain Polarity is polar
-32: Side Chain Polarity is basic polar
-32: Side Chain Polarity is basic polar
-33: Side Chain Charge is negative
-34: Side Chain Charge is neutral
-35: Side Chain Charge is positive
-36: Hydropathy Index(原始值除以5作归一化,直接使用连续值,未离散化作onehot)
-37: Molecular Mass(原始值减去均值再除以70作归一化,直接使用连续值,未离散化作onehot)
 此外,为了便于处理,我们统一将不同长度的氨基酸序列padding到700,保证比目前已知的最长氨基酸序列长,因此20000条氨基酸序列处理后可得到(20000 × 700 × 38)。
此外,为了便于处理,我们统一将不同长度的氨基酸序列padding到700,保证比目前已知的最长氨基酸序列长,因此20000条氨基酸序列处理后可得到(20000 × 700 × 38)。
# onehot for 21 amino acid & features
seqdict = {
"G": [[0, 22, 30, 34], -0.08, -0.88],
"P": [[1, 26, 30, 34], -0.32, -0.31],
"T": [[2, 27, 31, 34], -0.14, -0.25],
"E": [[3, 21, 29, 33], -0.7, 0.15],
"S": [[4, 27, 31, 34], -0.16, -0.45],
"K": [[5, 25, 32, 35], -0.78, 0.13],
"C": [[6, 28, 30, 34], 0.5, -0.22],
"L": [[7, 22, 30, 34], 0.76, -0.08],
"M": [[8, 28, 30, 34], 0.38, 0.18],
"V": [[9, 22, 30, 34], 0.84, -0.28],
"D": [[10, 21, 29, 33], -0.7, -0.05],
"A": [[11, 22, 30, 34], 0.36, -0.68],
"R": [[12, 25, 32, 35], -0.9, 0.53],
"I": [[13, 22, 30, 34], 0.9, -0.08],
"N": [[14, 23, 31, 34], -0.7, -0.07],
"H": [[15, 24, 25, 32, 33], -0.64, 0.26],
"F": [[16, 24, 30, 34], 0.56, 0.40],
"W": [[17, 24, 30, 34], -0.18, 0.96],
"Y": [[18, 24, 31, 34], -0.26, 0.63],
"Q": [[19, 23, 31, 34], -0.7, 0.13],
"X": [[20], -0.04, 0]
}
# onehot for 8 structure
secdict = {
" ": 0,
"E": 1,
"T": 2,
"S": 3,
"H": 4,
"G": 5,
"B": 6,
"I": 7,
}
table = [' ', 'E', 'T', 'S', 'H', 'G', 'B', 'I']
filepath = r"test.txt" #
n = len(open(filepath, 'r').readlines())
X_test = np.zeros((n, 700, 38))
Y_test = np.zeros((n, 700, 1))
i_num = 0
# preprocess
with open(filepath) as f:
for lines in f:
i_char = 0
for char in lines[:-1]:
indexlist = seqdict.get(char)
if indexlist == None:
indexlist = seqdict.get("X")
for index in indexlist[0]:
X_test[i_num][i_char][index] = 1
X_test[i_num][i_char][36] = indexlist[1]
X_test[i_num][i_char][37] = indexlist[2]
Y_test[i_num][i_char][0] = 1
i_char += 1
i_num += 1
模型介绍与训练
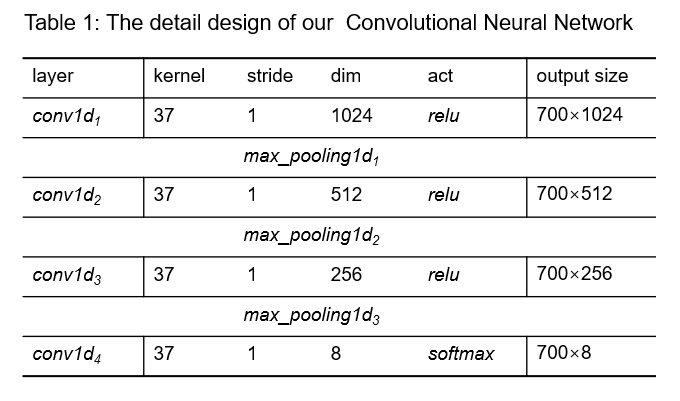 我们使用了一个朴素的四层卷积神经网络模型,每层的kernel_size均选择37,以一个较大的局部感受野模拟氨基酸序列的长程作用,使用较高维度的输出通道学习更多更复杂的特征,同时使用dropout和pooling防止过拟合。我们使用95%的数据用于训练,5%的数据用于验证,网络训练时的参数如下:
我们使用了一个朴素的四层卷积神经网络模型,每层的kernel_size均选择37,以一个较大的局部感受野模拟氨基酸序列的长程作用,使用较高维度的输出通道学习更多更复杂的特征,同时使用dropout和pooling防止过拟合。我们使用95%的数据用于训练,5%的数据用于验证,网络训练时的参数如下:










