科研经验
|
文献
|
实验
|
工
具
|
SCI写作
|
国自然
作者:Lucky King
转载请注明:解螺旋·临床医生科研成长平台
今天讲的文献题目是“
一组MicroRNA作为鉴定前列腺癌的诊断性生物标志物
”,这是一篇干湿结合的小文章,影响因子3.226(回复
170718
可下载文献,一周有效)。

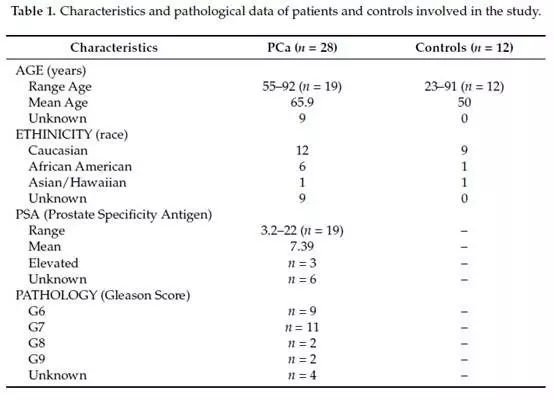
将患者(n=28)与对照(n=12)的病理数据、特征进行比较(表1)。
在诊断早期阶段、治疗前阶段,给患者取血。(Blabla。。。一大段临床病人筛选,此处省略一万字母)。
收集血液,从样本材料中提取miRNA。HTS数据显示,在miRBase中存在2588个miRNA,采集的样品中处于可检测到水平的miRNA约550个。为了更好地完善、不遗漏miRNA名单,使用Benjamini-Hochberg方法对p值进行调整,从而产生了False检测率、FDR值。FDR值表示使用广义线性化模型可能的错误检测率。
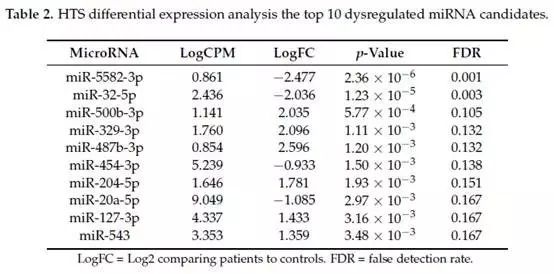
为了在HTS数据库中筛选出尽可能多的miRNA,FDR值<0.2的都被选中。FDR值为0.2意味着:选定的miRNA中20%可能是假阳性。作者后期还将使用qRT-PCR进行验证,所以初期使用HTS数据库时多选择了些miRNA。HTS数据库显示,差异表达失调的top 10 miRNA见表2。
由于miR-5582-3p和miR-543引物难合成,且样本中丰度低,所以舍弃了。另外miR-500b-3p其丰度也较低,舍弃。最终确定7种microRNA进行下步分析。
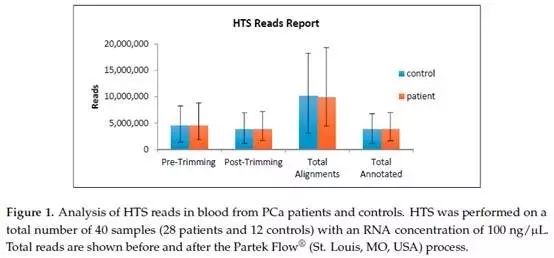
使用生物信息学分析,患者和对照组的HTS总读数没有显著差异,表明来自正常组和患者组的血液中含有相似量的总miRNA(图1)。作者认为,miRNA总量的相似可以证实某些miRNA的差异表达不是由于文库大小的差异引起的。
使用R程序,Trimmed Mean of M-values加权截断均值(TMM)法进一步处理原始数据。随后通过qRT-PCR证实了TMM方法对miRNA测序结果正常有效。
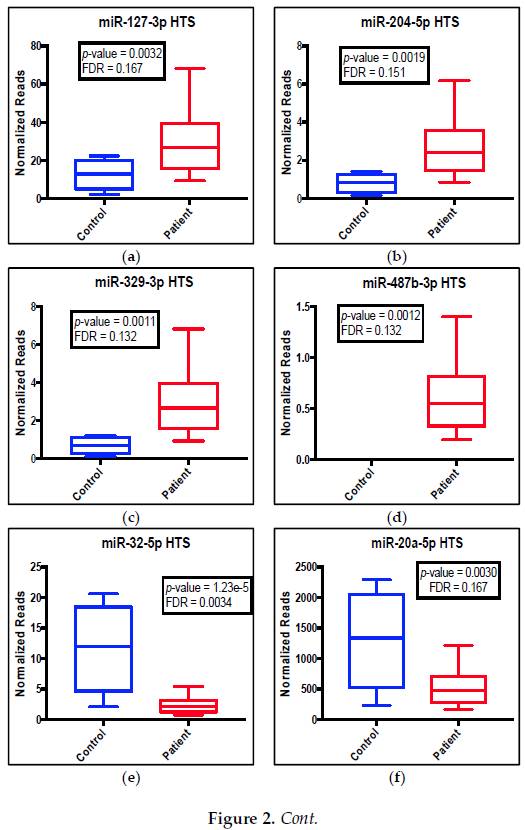
由HTS差异表达分析表明:七个失调的miRNA,对照组和患者组之间的归一化读数差异很大(图2 a-g)。患者血液样品中4种miRNAs(miR-127-3p、miR-204-5p、miR-329-3p、miR-487b-3p)上调(图2 a-d),而3种miRNA(miR-32-5p、miR-20a-5p、miR-454-3p)被下调(图2e-g)。
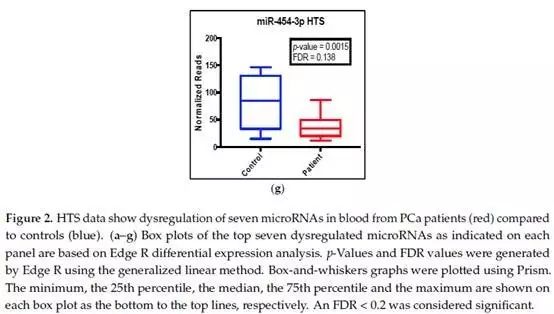
为了确认HTS数据的准确性可用性,使用qRT-PCR进行验证。以确保miRNA表达异常是由于真正的生物学变异而不是技术性错误。(原文过于洋气&啰嗦,就是验证)。
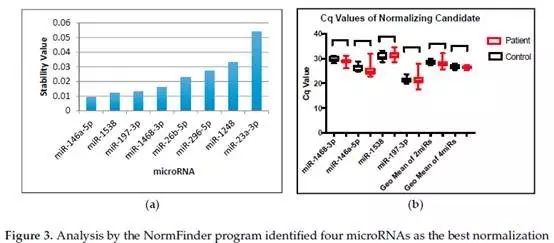
使用 NormFinder (NormFinder software)程序分析结果,该程序检查组内和组间差异以确定每个miRNA的稳定性值。值越低,越好。
NormFinder程序选择出8个稳定表达的miRNA:miR-146a-5p、miR-1538、miR-197-3p、miR-1468-3p、miR-26b-5p、miR-296-5p、miR-1248和miR-23a-3p(图3a)。
NormFinder筛选建议:单一最佳选项是miR-146a-5p。当没有突出的单一选项时,NormFinder建议使用miRNA组合来增加可靠性,并减少组内和组间差异性。
由于前四个miRNAs(miR-146a-5p、miR-1538、miR-197-3p、miR-1468-3p)均显示出非常接近的稳定性值,将每个miRNA的值、Top2(miR-146-5p和miR-1538)组合平均值、所有top 4的miRNA组合的平均值进行比较。
Top2组合显示组内变化减少,特别是对于患者组。Top4 miRNA组合的平均值,在对照组和患者组之间均显示出更小的内部变化(图 3b最后一组)。因此,选择top4 miRNA组合作为标准化用于下步qRT-PCR分析。
(这一大段是方法论,证明方法有理有据有效,可用于后续。这里的一堆miRNAs和前列腺癌一毛钱关系都木有。)
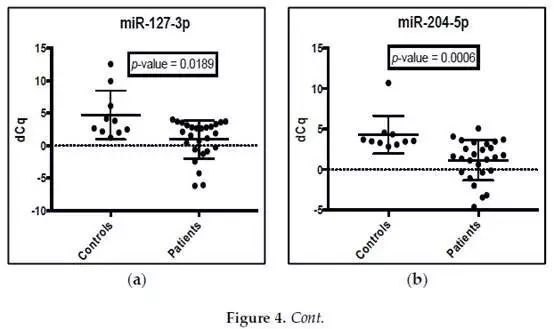
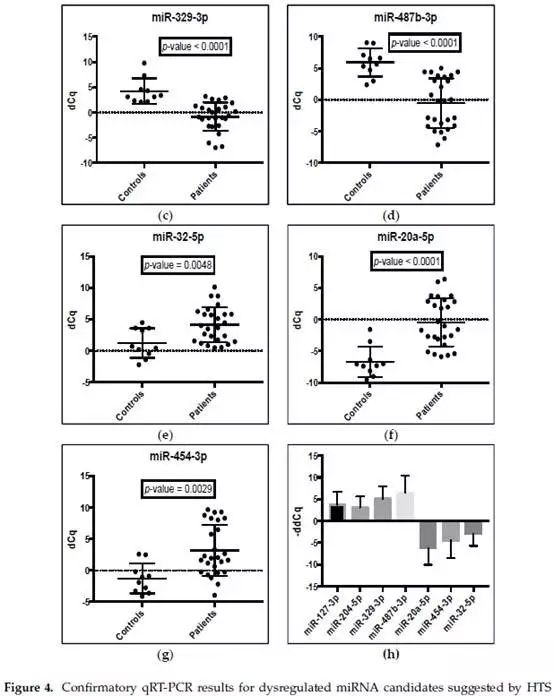
根据qRT-PCR结果,与对照组相比,miR-127-3p、miR-204-5p、miR-329-3p、miR-487b-3p在患者中上调(图4 a-d);而miR-32-5p、miR-20a-5p、miR-454-3p被下调(图4 e-g)。
qRT PCR结果与HTS数据分析结果一致,证实PCa患者中所有7种microRNAs均失调。
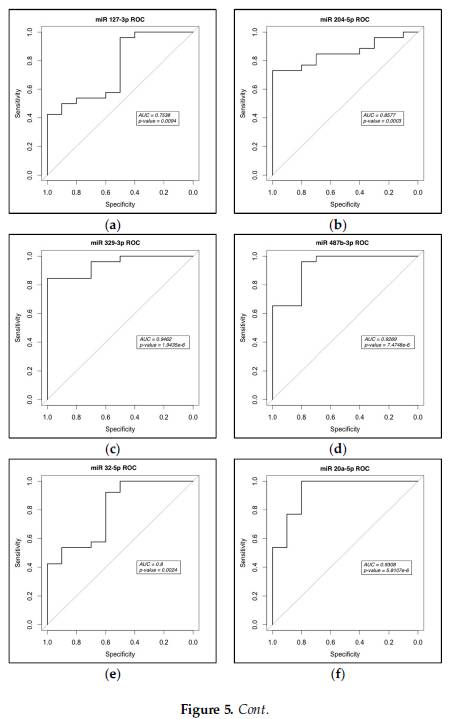
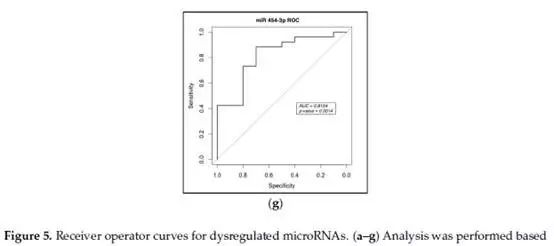
为了进一步评估七种microRNA是否可以作为良好的PCa生物标志物,基于qRT-PCR数据绘制ROC曲线。每个miRNA的ROC曲线如图5所示。
在ROC分析中,曲线下面积(AUC)量化值越高,区分PCa患者和对照组的效果越好。通过ROC分析,现阶段使用的PCa生物标志物PSA的AUC值为0.678(金标准),所以将0.678作为区分PCa与无癌症的临界值。这七种miRNA的AUC值范围从0.7538(miR-127-3p)到0.9462(miR-329-3p),均优于已报道过的PSA(Figure5 a–g)。
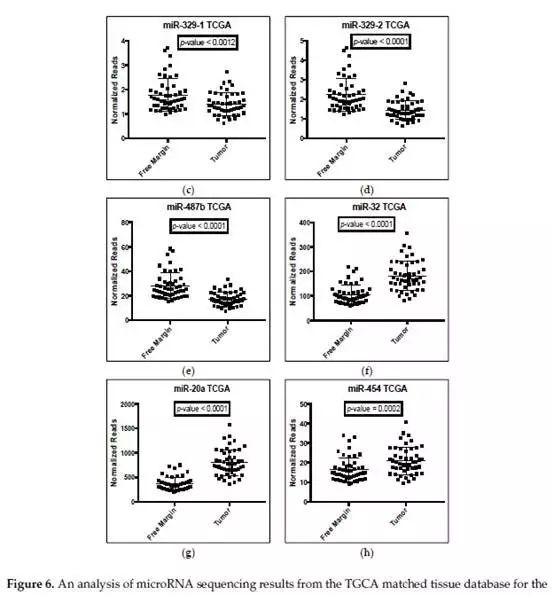
通过对TCGA中数据的分析,将血小板与肿瘤组织中的表达水平进行比较。TCGA中miRNA序列数据是茎环转录本而不是成熟链,研究中的七个miRNA数据是基于miRBase,主要为成熟链。
因此,这七种成熟miRNAs的表达与其茎环前体的丰度成正比。成熟的miR-127-3p、miR-204-5p、miR-487b-3p、miR-32-5p、miR-20a-5p和miR-454-3p衍生自前体miR-127、miR-204、miR -487b、miR-32、miR-20a和miR-454。成熟的miR-329-3p来自两个前体,miR-329-1和miR-329-2。
PCa组织中每种前体的表达与其无疾病的癌旁组织相比显示出显著失调,并且失调方向与文献结果一致(图6)。然而,肿瘤组织中每个miRNA的失调模式与血液样本中观察到的相反。例如:miR-127、miR-204、miR-329-1、miR-329-2和miR-487b都在PCa组织中上调,这表明它们的主要成熟链(miR-127-3p、miR-204-5p、miR-329-3p、miR-487b-3p)也被上调。
然而,这四种miRNAs在血液样本中显示被下调。对于血液中下调的三个miRNA(miR-32-5p、miR-20a-5p、miR-454-3p)观察到逆相关。再次,其前体转录物和可能主要的成熟miRNA产物在TCGA数据中上调。
做了一堆分析,开始7个最后还是7个miRNAs,得出结论是一组在一起方可预测诊断前列腺癌。真要用于临床诊断筛查,这作者是要给医院创收啊,直接翻7倍。
本文是一篇典型的,干分析+湿试验,干湿结合的文章。试验不多,仅有RT-PCR,安生做也就不到一个月的劳力。3.226的分数足够不少学校的博士毕业了吧。非要写路线的话:

本文的重点&难点不在于思路、idea,而是数据库&软件的使用:
-
常见的如TCGA;
-
miRBase(microRNA序列数据库www.mirbase.com);
-
high throughput screening,高通量筛选HTS;
-
NormFinder software;
-
ROC曲线。
数据库&软件教程PPT,度娘上现成的。相信有心人,已经在路上了。
福利:
长按识别下方二维码添加“文献菌”为好友,即日起至8月31日,春季班学员立刻获得【文献精读 秋季班】
19.9元
购买权益。(原价99元)

如果想看全部的文献精读课,可以扫下面的二维码购买课程录播(
课程有效期至2018年12月31日
),想怎么看就怎么看。

点击底部“
阅读原文
”,即可查看课程详情





