
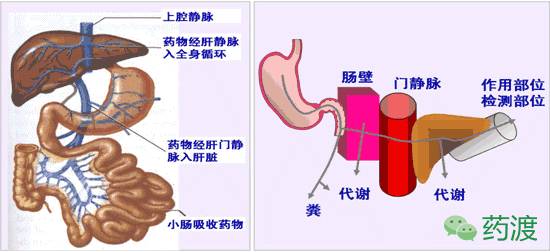
所有口服药物的吸收须透过胃肠壁,然后进入门静脉,经门静脉系统进入肝脏,在肝药酶、胃肠道酶和微生物的联合作用下进行首次代谢,使进入全身药量减少。有些药物几乎无代谢作用发生,有些则在胃肠壁或肝脏内被广泛代谢、消除,发生首过作用,称为首过效应(first pass effect),亦称为首过代谢(first pass metabolism)。发生首过代谢是因为人的解剖结构和生理作用决定了有这种现象。药物经消化道吸收后,经门静脉进入肝脏,而肝脏有P450系统(特异性比较低的一群氧化酶),对很多药物都有代谢作用。
首过代谢应该理解为人体的一种生理现象,不是药物特有的现象。其生理意义在于保护人体免受经消化道吸收入体内的化学物质的作用(正如一般认为的肝脏是一个解毒器官)。这种生理作用不是针对药物而存在的,而是应对自然环境中毒物的防御屏障。只是在药理学研究时,发现这种现象,把它命名为首过代谢。其次就是经消化道吸收药物和其它进入人体的化学物质一样可能要经历首过代谢。
到目前为止还没有研究证据说明这种生理作用对人类有什么危害。首过代谢明显的药物,口服生物利用度低,和注射给药比较,口服必须用更大剂量才能达到相当的有效血药浓度。肝清除药物能力的改变能显著影响血药浓度。
药物的“首过效应”一般可分为三种:
①胃肠道首过效应,即口服药物经过消化道时,在胃肠道代谢酶的作用下,可能发生各种代谢反应,导致部分药物在胃肠道中代谢失活,结果使吸收进入体内的原型药物相应减少。
②肝首过效应,即从胃肠道吸收的药物经过肝门静脉进入肝脏,部分药物被肝药酶代谢转化,或与组织成分结合,或随胆汁排出,使进入体循环的原型药物量减少。
③肺首过效应,此类药物极少,如氯仿等。
口服药物吸收进入小肠或者大肠毛细血管的任何外来分子,在进入身体的其它部位之前,都必须通过门静脉经过肝脏到达体循环。只有那些经过淋巴系统或者远端直肠吸收的药物才能绕过肝脏直接进入体循环。口服药物的主要吸收部位在小肠。小肠大体上可以分为十二指肠、空肠、回肠三部分,这三部分组织学特点基本一样,但肠上皮细胞层是药物的吸收和首过代谢的重要部位。
口服给药的疗效比非胃肠道给药要低得多,造成这种现象的原因包括药物分子在胃肠道环境下的化学降解、药物从制剂中的释放不完全、肠渗透性差、通过胃肠道时药物与肠黏膜的接触时间不足、以及通过肠道及肝脏时的首过代谢等。其中广泛的首过代谢是药物口服生物利用度低、疗效差的主要原因,限制药物在胃肠道释放的因素可以通过适当的剂型加以克服,而首过代谢则是获得最佳生物利用度不可避免的障碍。一般认为有较高首过代谢的口服药物,其生物利用度往往也存在较大的个体差异。
药物吸收的最佳部位是在近端小肠的绒毛和微绒毛。形成肠腔内表面屏障的上皮细胞内含有相当多的药物代谢氧化酶、结合酶、水解酶等。外来药物分子可以通过扩散或者载体转运经过成熟的肠上皮细胞的质膜进入毛细血管,但同时要经历胞浆内酶代谢屏障。若粘膜上皮细胞内的酶活性足够高,则肠首过代谢可达到100%的提取率。
在胃肠道粘膜的上皮细胞中可以检测到肝内存在的很多药物代谢酶,主要有CYP450、葡萄糖醛酸转移酶、磺基转移酶、N-乙酰基转移酶、谷胱甘肽S-转移酶、酯酶、环氧化物水化酶和乙醇脱氢酶等。
CYP450
在胃肠道粘膜微粒体中P450酶含量随解剖学部位的不同而不同。在食管、胃、结肠仅发现有极微量的CYP3A、2E1、2D6、1A2、2C,而在小肠中却含有较高水平的CYP3A、2D6、2C等,小肠微粒体中CYP3A和CYP2D6的平均含量分别是肝内相应水平的50%和10%~20%,小肠中CYP3A的表达水平高于其它血液丰富的肝外组织,如肾、肺等。在小肠中,近端粘膜(十二指肠到空肠)CYP450含量高于远端(回肠)粘膜,从回肠到结肠,CYP450的含量进一步下降。并且由于从近端到远端粘膜质量也在下降,实际上粘膜中CYP450总含量的下降应比微粒体内含量的下降趋势更大。
CYP3A4是人体胃肠道中占主导地位的CYP450,是小肠粘膜上皮细胞中微粒体CYP450的主要形式。在Paine等人进行的二十例人体全小肠的研究中发现,微粒体CYP3A4的表达沿整个小肠是延续的,但其含量随部位而变化。其平均含量在十二指肠、空肠、回肠中分别是31 (<2~91)、23 (<2~98)、17 (<2~60) pmol/mg。
CYP3A4存在于长绒毛成熟上皮细胞内,其最高含量出现在吸收表面附近,即在刷状缘膜之下,但并不存在于隐窝细胞中。肠上皮细胞内的CYP3A4与肝内CYP3A4在结构与功能上是一致的,另外,一些CYP3A4底物在小肠和肝内的代谢产物和代谢动力学特征也是一致的。
CYP3A5在人小肠与肝内的表达相似,呈现多态性特点。它在大多数个体的肠上皮细胞中均不是主要代谢酶,CYP3A5的结构、功能和其对首过代谢的贡献到目前为止尚未完全弄清楚。
人胃中CYP3A5的水平虽然比小肠中低得多,但却是主导性的,人体结肠中CYP3A5相对于CYP3A4也是占主导地位的。CYP2C亚族在肠上皮细胞中表达水平居第二位。CYP2J最早是从兔子小肠分离得到的,其亚型为CYP2J1,人体内为CYP2J2,在人体小肠、肝、肾、心等均有表达,并且可能在花生四烯酸的氧化代谢中起作用。CYP1A1主要存在肝外组织。
P-糖蛋白
对于CYP3A底物的吸收还必须要考虑另外一个因素,即发生在绒毛顶端上皮细胞顶端膜上的外排作用,外棑转运体有P-糖蛋白(P-glycoprotein,P-gp)和多药耐药相关蛋白2 (MRP2)。
P-gp与MRP2可使药物外排至肠腔,可有效地降低药物在肠上皮细胞内的停留,并泵出很多外来异物如环孢素、紫杉醇、地尔硫、地塞米松、利多卡因、红霉素及蛋白水解酶抑制剂等。
在小肠中,P-gp几乎是专属性地分布于肠绒毛成熟上皮细胞顶端表面的刷状缘膜内。在该部位将外来异物从细胞质内泵出到细胞外,从肠上皮细胞内泵回到肠腔。如果P-gp的活性能将药物吸收延迟到小肠的远端或大肠,由于肠道远端的吸收表面积降低、且细胞间连接更紧密,这可能会较大地降低口服生物利用度。
口服药物是通过肠腔内扩散到达代谢部位,药物进入顶端上皮细胞及到达毛细血管的扩散转运与细胞内的代谢消除相互竞争;口服吸收后,药物进入顶端上皮细胞的过程在整个肠道是不均匀的,并非所有的代谢部位都可以在任何时候暴露于药物,由于粘膜中酶的含量不同,内在代谢清除率随小肠的部位不同而变化;代谢酶在结肠和胃的表达水平很低,所以药物从幽门到回盲瓣的平均运行时间就成为一些药物肠首过代谢的限制因素,特别是对缓释剂而言。采用放射性标记的纤维素内镜探针测得健康志愿者体内平均肠运行时间为3.65h。药物对绒毛内代谢酶的有限暴露时间将影响小肠对药物的最大转化率。此外,肠运行时间可能也是肠首过代谢个体间和个体内差异的来源之一。
肠首过代谢是指口服药物首次经过肠壁时被代谢消除的过程,肠系统提取是指吸收入血的药物经动脉血分布到达肠道时被代谢消除的过程。肠对药物的首过提取率可能超过肠的系统表观提取率。肠系统表观提取率主要受系统动脉血流量中灌注绒毛上皮细胞或代谢隔室的血流量的主导;相反,对于肠首过提取,口服剂量所有吸收的药物在进入体循环之前,都要通过代谢隔室。在肝移植患者无肝期进行的咪达唑仑动力学研究结果证实了这个理论观点。在静脉注射咪达唑仑时测得肠平均提取率为8.2%±11.5%,在十二指肠内给药收平均首过提取率高达43.0%±18.1%。另外,肠首过提取率是以质量比为指标的,而肝首过提取率是以通过器官的血药浓度的差异为指标。除血流量外,血浆蛋白结合可以限制药物的传递,因此会影响药物从系统血液循环的提取,对首过提取也会有一些影响。
当比较肠系统提取和首过提取时,应该注意肠系统提取时动脉血中的药物是以相同的血药浓度到达粘膜代谢酶部位,而肠首过提取时口服的药物仅能到达一部分肠代谢部位,并且其浓度也是不同的。可以假设在吸收相上皮细胞内未结合型药物浓度要大大高于动脉内未结合型药物浓度,在吸收相肠首过代谢达到饱和的可能性要远大于消除相及其末端。
肝首过代谢,又称“肝首过效应”(liver first pass effect):透过胃肠道生物膜吸收的药物经过肝门静脉入肝后,在肝药酶作用下药物可产生生物转化。药物进入体循环前的降解或失活称为“肝首过代谢”或“肝首过效应”。
肝首过效应愈大,药物被代谢越多,其血药浓度也愈小,药效会受到明显的影响。
自消化道吸收的药物经门静脉系统进入肝,由于肝脏的首过作用,最终导致出肝药物浓度低于入肝药物浓度,入肝浓度和出肝浓度的差值(CA-CV)与入肝浓度(CA)之比即为肝提取率(hepatic extraction ratio EH)。
肝脏由干细胞组成,并有丰富的血管网。人肝约有25亿个干细胞,5千个干细胞组成一个小叶,因此人肝的肝小叶总数约有50万个,肝细胞由许多复杂的维系结构组成。肝脏主要有代谢功能、胆汁生成和排泄功能、解毒功能、血液凝固功能、免疫功能,以及参与人体血容量、热量的产生和水、电解质的调节等功能。内质网是干细胞中与药物代谢密切相关的细胞器,分为粗面内质网和滑面内质网两种。粗面内质网是蛋白质合成的重要场所,滑面内质网广泛分布于干细胞质内,是粗面内质网的2.5~3.0倍。
绝大多数药物代谢都是在肝细胞内药物代谢酶的催化作用下发生的,少数药物代谢反应可以自发进行,如在体液环境下发生水解等反应,这些反应不需要酶催化。肝内CYP450、葡萄糖醛酸转移酶、磺基转移酶及其他药物代谢酶都是很丰富的。从首过代谢的角度,CYP450是最重要的一类肝药酶。据估计肝内含有大约25000nmol的CYP450亚族及其多种同工酶,其中对于首过代谢具有重要意义的有CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4。CYP3A4是最常见的同工酶,由于其在胃肠道粘膜和肝中均有表达、丰度最大、底物特异性较广等而显得尤其重要。值得注意的是,从60例人肝微粒体得到的数据可见,CYP450中各种同工酶的含量于其对代谢的贡献并不一致,如CYP3A在人体肝细胞CYP450中约占29%,但约有50%的药物代谢反应受CYP3A介导,CYP2D6约占人肝CYP450的1.7%,但参与的代谢反应却仅占30%(如下图)。
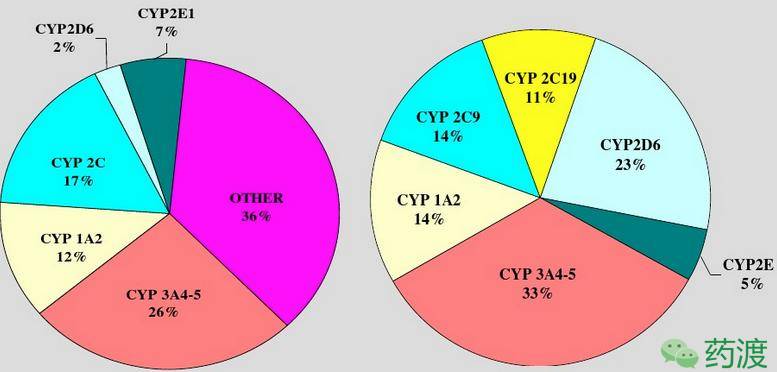
人肝微粒体内CYP450同工酶的相对丰度和对药物代谢的相对贡献示意图
随着对药物代谢酶反应机制的认识,整体代谢可采用在体探针药物的代谢来表征体内代谢酶的活性,找出药物代谢与某些代谢酶活性的相关性,在给药前后给予探针药物可以反映药物对某些酶的诱导或抑制作用。清除率常作为药物代谢能力的指标,对于主要经过肝代谢的药物而言,该参数可直接反映肝代谢能力,如安替比林。还有些药物选择性地经某种同工酶代谢,其清除率则可作为该同工酶的活性指标。如咖啡因、茶碱主要经CYP1A代谢,美芬妥英主要经CYP2C9代谢。
首过效应常使药物的生物利用度明显降低,有些药物甚至由于首过效应强烈,大部分被代谢而失去活性,导致无法口服给药。那么为了提高生物利用度,降低药物首过效应,目前常用的避免药物首过代谢的方法主要有:
①改变给药途径。如口服硝酸甘油受首过效应代谢90%左右,故疗效差,采用绕过首过效应,而舌下含用,直接由舌下粘膜吸收进入血液,药物破坏较少。某些药物也可采用经肛门灌肠或用栓剂按被动转运方式吸收,由于接触面小,吸收量较口服为少,但由于吸收途径不经肝脏,药物破坏较少,作用较快。
②选择适当的给药时间,如进食时服药能减少药物在肝脏的代谢,提高生物利用度。
③首次剂量加倍,能抵消肝药酶代谢作用,有些药也可采用首次一日剂量一次服用。
参考文献:
[1]李光华,口服药物吸收的研究进展,天津药学,2014年第26 (2),69~71.
[2]实用药理学[M]. 贵州人民出版社 ,王焕斗 主编,1982
[3]杨宝峰主编.药理学第八版.人民卫生出版社,2013-3-1










