HiC-Pro是一个高效率的Hi-C数据预处理工具,能够应用于dilution Hi-C, in situ Hi-C, DNase Hi-C, Micro-C, capture-C, capture Hi-C 和 HiChip 这些数据中。
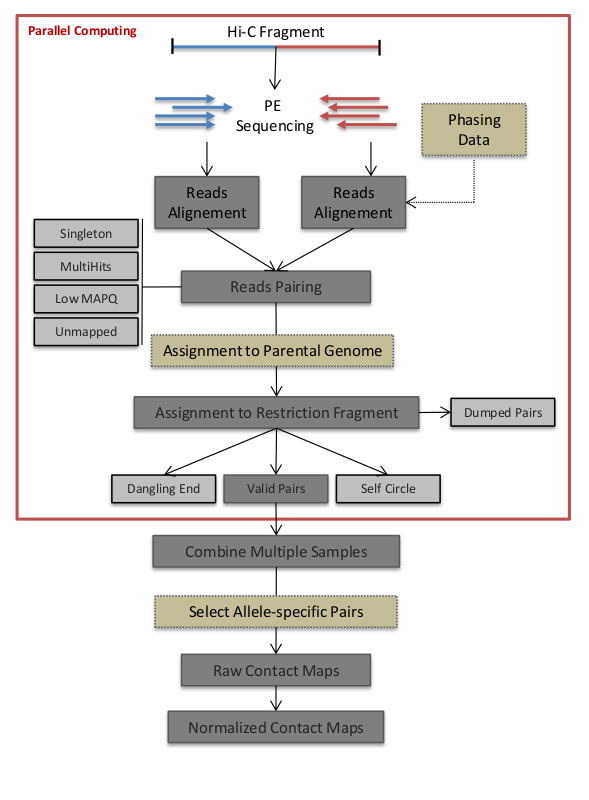
HiC-Pro的工作流程如下, 简单的说就是先双端测序各自比对,然后进行合并,根据合并的结果筛选有效配对。之后有效配对用于构建contact maps.
安装方法
HiC-Pro依赖于如下的软件
-
Bowtie2(>2.2.2), 用于序列比对
-
Python2.7, 并安装 Pysam, bx-python, numpy, scipy
-
R, RColorBrewer + ggplot2
-
samtools > 1.1
-
GNU sort, 支持 -V, 按照version进行排序
为了保证环境的干净,我用conda进行了一个环境进行安装
conda create -y -n hic-pro python=2.7 pysam bx-python numpy scipy samtools bowtie2
conda activate hic-pro
以2.11.1版本为例进行介绍,最新的版本在https://github.com/nservant/HiC-Pro/releases检查
wget https://github.com/nservant/HiC-Pro/archive/v2.11.1.tar.gz
tar -zxvf v2.11.1.tar.gz
cd HiC-Pro-2.11.1
因为我希望把HiC-Pro安装到
~/opt/bisofot
下,所以我需要修改当前目录下的
config-install.txt
中的PREFIX部分,
PREFIX = /home/xzg/opt/bisofot
如果服务器支持任务投递,可以修改CLUSTER_SYS部分, 设置为TORQUE, SGE, SLURM 或 LSF,
我的miniconda的安装目录是
~/miniconda3
, 所以hic-pro环境的实际路径是
~/miniconda3/envs/hic-pro
make configure
make install
安装结束之后,
/home/xzg/opt/bisofot
文件夹下就出现了
HiC-Pro_2.11.1
,之后的软件调用方式为
~/opt/biosfot/HiC-Pro_2.11.1/bin/HiC-Pro -h
如果没有出现Error 就说明安装成功了。
处理流程
让我们新建一个项目文件夹,以一个测试数据集为例进行介绍。
下载测试数据并解压缩,该数据来自于Dixon et al. 2012 , 使用HindIII 进行酶切
mkdir -p hic-pro && cd hic-pro
wget https://zerkalo.curie.fr/partage/HiC-Pro/HiCPro_testdata.tar.gz && tar -zxvf HiCPro_testdata.tar.gz
之后将测序结果移动或者软连接到fastq文件夹下
mkdir -p fastq
mv test_data/* fastq
ls fastq
创建注释文件
为了处理原始数据,HiC-Pro需要三个注释文件
其中BED文件和table文件必须要放在
HiC-Pro_2.11.1/annotations
目录下,该文件夹下已经有了人类hg19和小鼠mm10。我们以GRCh38为例, 介绍如何创建这三个注释信息
cd ~/opt/biosfot/HiC-Pro_2.11.1/annotation
wget ftp://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
gunzip Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
~/opt/biosoft/HiC-Pro_2.11.1/bin/utils/digest_genome.py -r HindIII -o GRCh38_HindIII.bed Homo_sapiens.GRCh38.dna.primary_assembly.fa
seqkit fx2tab -nl Homo_sapiens.GRCh38.dna.primary_assembly.fa | awk '{print $1"\t"$2}' > GRCh38.chrom.size
bowtie2-build --threads 20 Homo_sapiens.GRCh38.dna.primary_assembly.fa GRCh38
配置HiC-Pro
拷贝HiC-Pro的配置文件到项目文件夹下
cp ~/opt/biosfot/HiC-Pro_2.11.1/config-hicpro.txt .
修改配置文件config-hicpro.txt
N_CPU = 80
BOWTIE2_IDX_PATH = /home/xzg/opt/biosfot/HiC-Pro_2.11.1/annotation
REFERENCE_GENOME = GRCh38
GENOME_SIZE = GRCh38.chrom.size
GENOME_FRAGMENT = GRCh38_HindIII.bed
LIGATION_SITE = AAGCTAGCTT
对于LIGATION_SITE,不同酶切位点对应的序列为HindIII(AAGCTAGCTT), MboI(GATCGATC) , DpnII(GATCGATC), NcoI(CCATGCATGG)。
我们要修改的参数其实就是上面几个。当然该配置文件还有许多参数可以修改,具体见https://github.com/nservant/HiC-Pro/blob/master/doc/MANUAL.md
运行如下代码,启动分析项目
~/opt/biosoft/HiC-Pro_2.11.1/bin/HiC-Pro -i fastq -o results -c config-hicpro.txt
HiC-Pro会新建一个工作目录,results, 之后会遍历fastq目录,寻找其中的fastq文件,将其软连接到results下的rawdata, 之后就开始用bowtie2比对以及后续的分析。
测试数据代码运行到
Run ICE Normalization
就中断了,可能是用的参考基因组和原来的教程(hg19)不一样, 不过我自己的数据集是没有问题的。
最后的结果如下:
$ tree -L 2 results
results
├── bowtie_results
│ ├── bwt2![]()
│ ├── bwt2_global
│ └── bwt2_local
├── config-hicpro.txt
├── hic_results
│ ├── data
│ ├── matrix
│ ├── pic
│ └── stats
├── logs
│ ├── dixon_2M
│ └── dixon_2M_2
├── rawdata -> /home/xzg/project/Tutorial/hic-pro/fastq
└── tmp
一个关键的结果就是data文件里的以
.validPairs
结尾的文件,有7+1列,
read_name chr_reads1 pos_reads1 strand_reads1 chr_reads2 pos_reads2 strand_reads2 fragment_size [allele_specific_tag]
此外,HiC-Pro提供了一个脚本用于将输出的allValidPairs转成JABT的输入
~/HiC-Pro_2.11.1/bin/utils/hicpro2juicebox.sh \
-i hic_results/data/dixon_2M/dixon_2M.allValidPairs \
-g /home/xzg/opt/biosfot/HiC-Pro_2.11.1/annotation/GRCh38.chrom.size \
-j ~/opt/biosoft/juicer/scripts/common/juicer_tools.jar
最终会输出一个以.hic结尾的文件。
参考资料
-
官方手册:https://github.com/nservant/HiC-Pro/blob/master/doc/MANUAL.md
-
Servant N., Varoquaux N., Lajoie BR., Viara E., Chen CJ., Vert JP., Dekker J., Heard E., Barillot E.
HiC-Pro: An optimized and flexible pipeline for Hi-C processing. Genome Biology 2015, 16:259 doi:10.1186/s13059-015-0831-x




