为什么要进行培养基模拟灌装试验?
法规要求
生产工艺自身要求
培养基模拟灌装试验的法规要求
FDA CGMP 无菌工艺药品指南(2004)
应当使用微生物培养基替代产品来验证无菌工艺操作。这种工艺模拟,又称为培养基模拟分装。
EU GMP 附件1 无菌药品的生产(2008)
无菌工艺验证应包括一个使用营养培养基工艺模拟试验(培养基分装)。
2010版GMP附录一无菌药品:
第四十七条 无菌生产工艺的验证应包括培养基模拟灌装试验。
应根据产品的剂型以及培养基的选择性、澄清度、浓度和灭菌的适用性选择培养基。应尽可能模拟常规的无菌生产工艺,包括所有对产品的无菌特性有影响的关键操作,及生产中可能出现的各种干预和最差条件。
培养基模拟灌装试验的首次验证,每班次应连续进行3次合格的试验。空气净化系统、设备、生产工艺及人员重大变更后,应重复进行培养基模拟灌装试验。培养基模拟灌装试验通常应按生产工艺每班次半年进行1次,每次至少一批。
来自生产工艺的要求……
工艺特点
对产品进行最终灭菌,是降低微生物污染风险的常用办法。但有大量药品不适用于最终灭菌而只能采用无菌生产工艺方法。
无菌生产工艺是制药领域中较难的工艺之一,确保产品全程无菌是该工艺最大的难点。
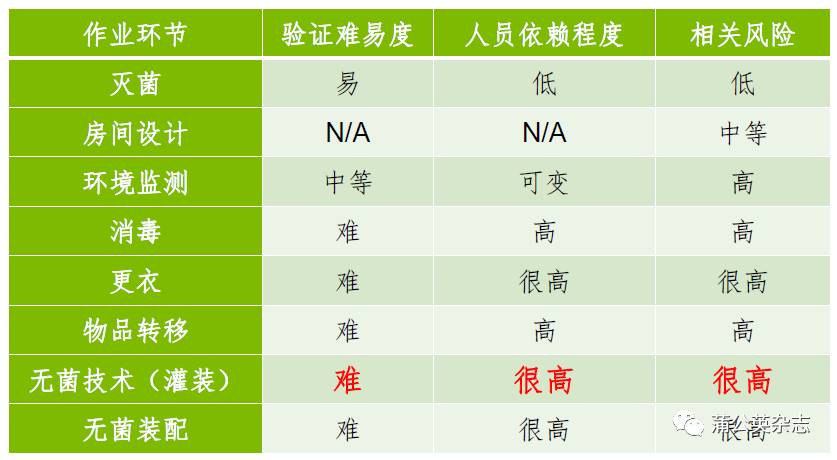
工艺环节与风险
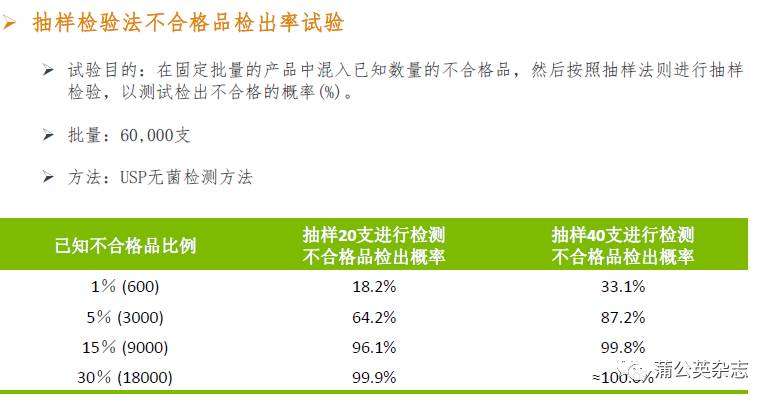
抽样检验法的缺陷
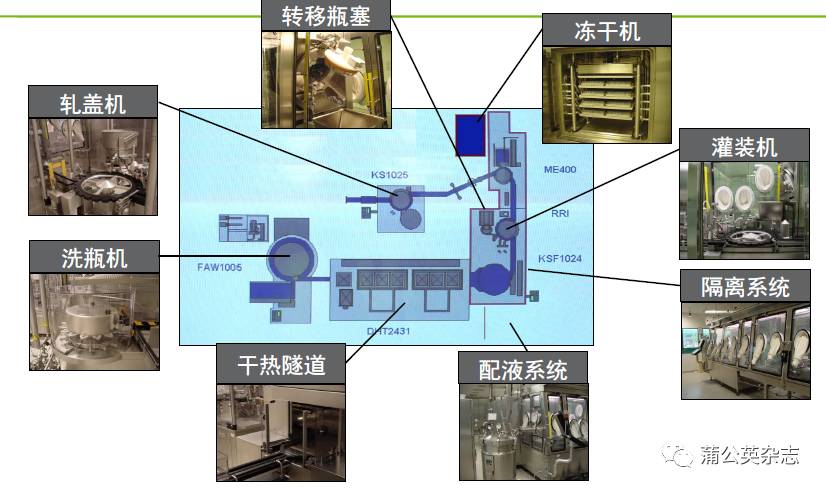
为什么要进行培养基模拟灌装试验
培养基模拟灌装的意义:
通过对生产工艺的无菌操作模拟,用培养基这种挑战性的产品来评价整个工艺系统(人员、设备、物料、程序和环境等)在某一个时间点的表现,以证明现有工艺系统的无菌保证水平能够保证产品的无菌性。
通过了培养基模拟灌装试验并不意味着工艺系统就始终处在相同的无菌保证水平。
无菌生产概况
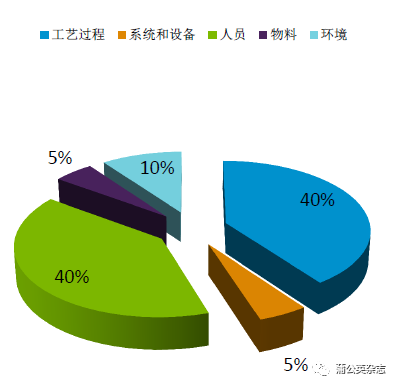
无菌保证的影响因素
内源性的影响因素
系统和设备
物料和中间体的质量
环境
工艺过程
外源性的影响因素
人员
培养基模拟灌装试验的基本条件
人员
设备
物料
方法
环境
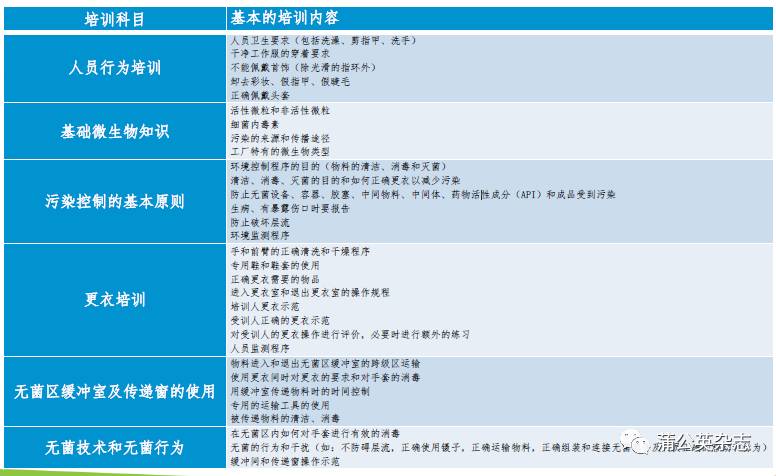
培养基模拟灌装试验的基本条件——人员
(
点击图片放大查看
)
进入无菌区从事与生产活动相关的操作人员和辅助人员须进行资格确认

无菌服是人与产品或环境之间的屏障,必须保持无菌服的完整性。
培养基模拟灌装试验的基本条件——设备
设计合理、功能完整的工艺设备也是必要的
.传统无菌操作模式
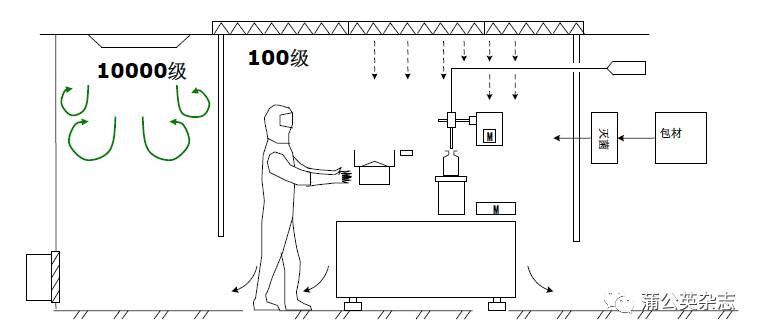
培养基模拟灌装试验的基本条件——物料
与正常工艺一致的物料
内/外包材
西林瓶/安瓿
胶塞
铝盖
合适的培养基的选择
适应广谱微生物生长,包括细菌和真菌
较好的澄明度,较小的粘度
易于除菌过滤
常用培养基:3%胰酪胨大豆肉汤培养基(TSB)
厌氧培养基仅在特殊情况下使用
培养基模拟灌装试验的基本条件——方法
初始验证
至少重复3次,以确保结果的重现性和有效性。
定期再验证
定期进行,评估无菌工艺的受控情况。
应综合考虑每一班次及班次变化过程中有代表性的活动及干预。
对人员的资格再确认应纳入到定期再验证中。
不定期再验证—变更及其评估
对产品或生产线的所有变更,均应根据书面的变更控制系统进行评估 。
如果通过评估认为此项变更会影响到无菌生产工艺产品的无菌保证水平,则需要进行增补性培养基模拟灌装试验。例如,设施或设备的变化、生产线配置的变动、人员重要变动、容器-胶塞系统变更。
其它如:环境监控结果异常、无菌检查结果阳性等。


灌装体积
每只容器的灌装体积不少于总容积的1/3
同样,灌装体积也不宜过大
既要考虑到瓶倒转或旋转时,培养基能充分接触到容器和密封件的内表面,又要保证容器内有足够的氧气支持微生物生长。
灌装数量
综合考虑统计学要求和实际生产批量。
FDA CGMP 要求>5000瓶,如果正常生产的批次量低于5000瓶,则培养基模拟灌装至少应为实际生产线的最大批次量。
CFDA GMP 2010 培养基模拟灌装容器的数量应足以保证评价的有效性。批量较小的产品,培养基模拟灌装的数量应至少等于产品的批量。
培养基模拟灌装试验的基本条件——环境
与正常工艺一致的环境
正常的清洁与灭菌程序,正常的环境监控方法
与正常生产一致的环境监控应该被正常执行
当环境结果不合格,但培养基模拟灌装试验通过时应分析说明环境对无菌工艺带来的潜在风险。
应包括悬浮粒子、沉降菌、浮游菌、人员监控和表面微生物监控等项目。
悬浮粒子
0.5 和 5.0 μm粒径的粒子应该被监控
Grade A 应该连续监控
Grade B 应该连续或定期监控
浮游菌
固定的取样量(例如: 1 m3 )
固定的或可移动的监控设备
提供量化的结果
沉降菌
应持续暴露在Grade A 的操作过程
固定的暴露时间(例如 2-4 小时)
提供量化的结果
表面微生物
使用接触碟或棉签获取表面微生物
固定的取样面积 (例如 20 cm2)
人员监控
监控无菌操作人员的服装和手套
监控点至少包括手套和前臂
培养基模拟灌装试验设计与执行
历史情况
风险分析
方案设计
挑战试验









