
作者:米粒儿(转载请注:解螺旋·医生科研助手)
PI3K通路的靶向药物在临床上可有效治疗乳腺癌。但是长期使用会产生耐药性,耐药现象常常伴随着雌激素受体(ER)介导的基因转录增强。深入理解PI3K和雌激素受体活性介导的耐药机制有助于解决PI3K通路靶向药物的耐药问题。今天介绍这篇论文发表主要关注PI3Kα抑制后,ER介导的基因转录增强机制。
文章标题:PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D(回复17405下载该文献)
本文研究发现在乳腺癌细胞模型和临床样本中,PI3Kα的抑制调节染色体状态。甲基转移酶KMT2D对于FOXA1, PBX1和 ER招募与活化是必需的。AKT1能够与KMT2D结合并将其磷酸化,该过程降低了甲基转移酶活性和ER的功能。
而PI3Kα的抑制可以增强KMT2D的功能。该研究阐明了表观遗传调节因子通过转录后修饰作用活化ER的机制,为ER阳性乳腺癌的治疗提供理论依据。
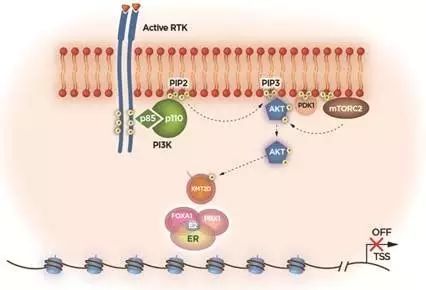
下面介绍一下作者是如何得到这些结论的。
PI3Kα抑制后,FOXA1和PBX1对于ER功能的活化是必需的
联合使用Chip-seq和Chip实验方法,发现在PI3Kα通路抑制后,呈现出不同的ER结合水平。并且发现在ER结合区域转录因子FOXA1和PBX1富集数量明显增加。随后采用Knockdown技术敲除FOXA1和PBX1后,进一步证明了FOXA1和PBX1在ER活化中的作用。
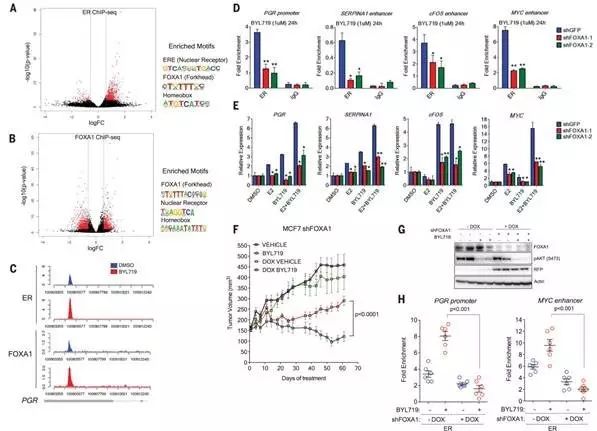
探讨PI3Kα抑制后,乳腺癌表观基因组染色质结构和ER-依赖的转录活性改变
采用ATAC-seq方法(检测染色质开放性以及TFs和染色质区域的相互作用),分别在细胞系(Fig.2A.B.C)和病人肿瘤组织(Fig. 2D.E)中,抑制PI3Kα后,染色质开放性和转录活性的改变。结果显示,PI3Kα通路抑制后,"染色质景观"改变导致ER-依赖的转录活化。
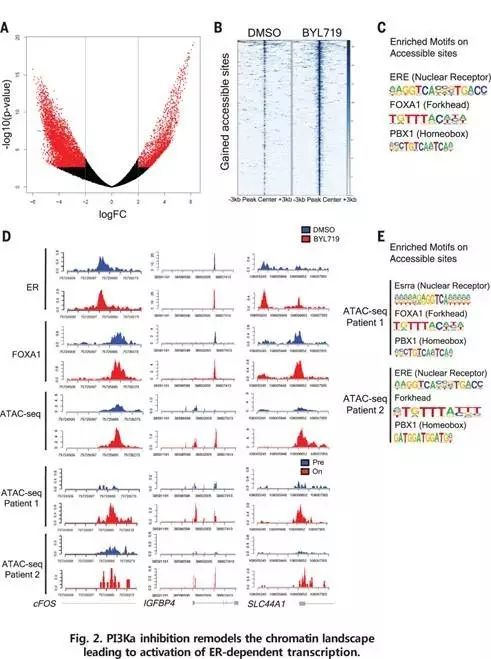
寻找PI3Kα抑制后,ER-依赖的转录调控因子
KMT2D是甲基转移酶,可与ER产生相互作用。因此选择KMT2D为研究对象,探讨KMT2D是否参与PI3Kα抑制后ER活性的调节。在PI3Kα抑制的细胞中,敲除KMT2D之后,检测ER,FOXA1,PBX1的招募以及ER靶基因的表达。
结果显示,KMT2D敲除之后这些TFs的结合能力显著下降,ER靶基因表达也显著下降(Fig. 3A和B)。
随后,为了验证KMT2D对肿瘤增殖的影响,作者采用体内实验检测敲除KMT2D之后,肿瘤体积的变化,发现敲除KMT2D之后,呈现出显著的抗肿瘤效应。当体系中同时敲除KMT2D和加入PI3Kα抑制剂后,抗肿瘤效应明显增强。
进一步的Chip-qPCR实验表明,KMT2D敲除之后,ER, FOXA1和 PBX1的结合作用消失。
以上实验结果表明,在ER阳性乳腺癌细胞中,靶向敲除KMT2D可增强PI3Kα抑制剂的敏感性。
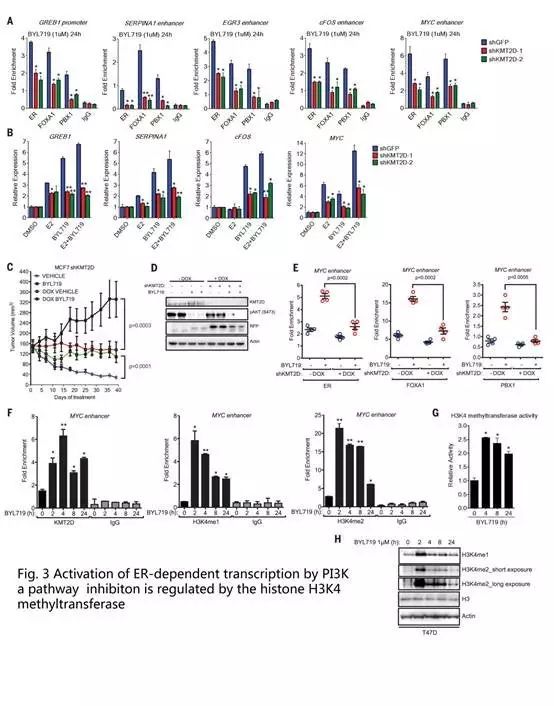
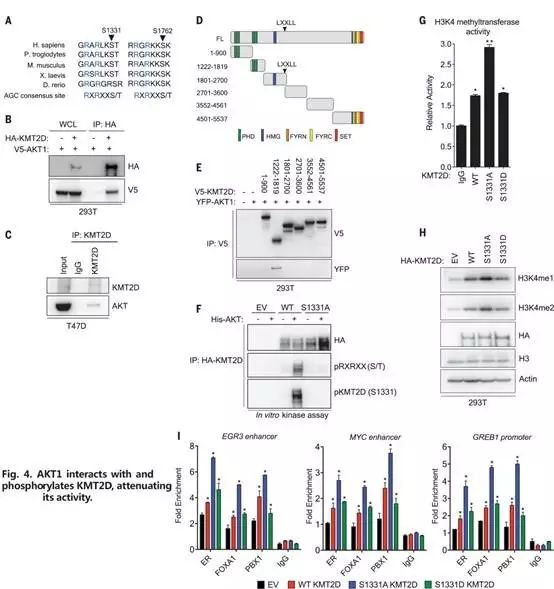
AKT1与磷酸化KMT2D相互作用后,降低其活性
PI3K通路的下游有很多不同的分子,通过观察AKT1和KMT2D的序列,作者发现二者可能存在相互作用。Co-IP实验也证明了这一结果,进一步的确定了KMT2D与AKT1相互作用区域。 此外,作者还发现KMT2D的S1331可被AKT1磷酸化,且磷酸化作用抑制KMT2D的甲基化水平,导致ER活性降低。

下边我们梳理一下本文的研究思路,研究流程见上图所示。本文能够发表在Science上的原因可总结为两点:
1、研究有很强的创新性和实用性,这是目前第一篇采用表观遗传学解释乳腺癌耐药机制的论文。研究结果为临床上解决乳腺癌耐药问题提供了依据。
2、研究采用了目前最新的检测技术,如ATAC-seq等。这些技术的应用保证文章数据的可靠性。
但是,大多数的研究者而言很难做到以上两点,那么我们应该如何借鉴这篇论文做相似的研究呢?
这篇文章究其根本是一篇研究表观遗传学的论文,表观遗传学的论文主要通过解释分子的表观修饰作用而阐述肿瘤表型的变化。
本文中ER活性,耐药两个"表型"的存在使得问题复杂化,而我们在研究中可以只保留耐药这一问题,研究内容只保留上图中的2和4即可。这样论文就变成了如下的模式:
1.PI3K抑制剂通过影响染色质开放性介导耐药
2.KMT2D的活性影响PI3K抑制剂的耐药
3.AKT1与磷酸化KMT2D相互作用后,降低其活性
参考文献:PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D
长按识别下方二维码添加文献菌为好友,直接提问和深入交流。

投稿邮箱: [email protected]
合作微信:helixlife6











