
【转载自:肖恩大侠】
“条条大路通罗马,每个靶点值万金。”每一个药物研发的靶点都可能是一个上十亿上百亿美金的重磅产品,当然,也可能是个研发的无底洞。本文简要汇总了目前在抗肿瘤药物靶点的一些基本概括、研究方向、主要热点问题与趋势等,供大家参考。此外,关于各靶点相关药物的产品、公司、市场分析则可以更多线下探讨。
导语
恶性肿瘤是危害人们生命健康的重大疾病,抗肿瘤药物的研发任重而道远。近年来,随着肿瘤生物学及相关学科的飞速发展,人们逐渐认识到细胞癌变的本质是细胞信号转导通路的失调导致的细胞无限增殖,随之而来的是抗肿瘤药物研发理念的重大转变。
研发的焦点正在从传统细胞毒药物转移到针对肿瘤细胞内异常信号系统靶点的特异性新一代抗肿瘤药物。不同于传统细胞毒药物选择性差、毒副作用强、易产生耐药性等特点,靶点特异性抗肿瘤药针对于正常细胞和肿瘤细胞之间的差异,达到了高选择性、低毒性的治疗效果,包括靶向酪氨酸激酶,血管新生,肿瘤细胞周期相关因子,组蛋白去乙酰酶抑制剂,微环境,肿瘤干细胞,肿瘤代谢异常等。
一、 靶向蛋白酪氨酸激酶(tyrosine kinase)
蛋白酪氨酸激酶是一类具有酪氨酸激酶活性的蛋白质,主要分布在细胞膜上,可分为受体型和非受体型,其功能都是催化ATP的磷酸基转移到下游蛋白的酪氨酸(Tyr)残基上,使其发生磷酸化。蛋白酪氨酸激酶是一个庞大的体系,目前已经发现了100多种酪氨酸激酶,分属20多个受体酪氨酸激酶家族和10个非受体酪氨酸激酶家族。
蛋白酪氨酸激酶在细胞信号转导通路中占据了十分重要的地位,调节着细胞的生长、分化、死亡等一系列生理生化过程。酪氨酸激酶的功能和肿瘤的发生、发展密切相关,超过50%的原癌基因和癌基因产物都是酪氨酸激酶,它们的异常表达通常导致细胞增殖调节发生紊乱,致使肿瘤发生。此外,酪氨酸的异常表达还与肿瘤的侵袭、转移、肿瘤新生血管生成以及肿瘤的化疗抗药性密切相关。
基于近年来在基因组学、分子和细胞生物学以及生物信息学等学科取得的重大进展,越来越多的酪氨酸激酶被认为是很有希望的抗肿瘤分子靶点。目前有超过20个分属不同家族的受体和非受体酪氨酸激酶被作为靶标进行抗肿瘤药物筛选,包括表皮生长因子受体(EGFR)、血管内皮细胞生长因子受体(VEGFR)、血小板衍生生长因子受体(PDGFR)、成纤维细胞生长因子受体(FGFR)、胰岛素受体(InsR)、Src、Abl等。靶向酪氨酸激酶的药物分为抗体类和小分子抑制剂。
1998年,Genetech公司和Roche联合开发的首个靶向HER2/neu的人源化单抗Herceptin被美国食品药物管理局(Food and Drug Administration,FDA)批准用于治疗某些HER2阳性的转移性乳腺癌。首个上市的小分子酪氨酸激酶抑制剂是特异靶向Bcr-Abl的Gleevec(Norvatis公司),已先后被FDA批准用于慢性髓样白血病(chronic myelogenous leukemia,CML)和胃肠道间质瘤(gastrointestinal stromal tumors,GIST)的治疗。从1998年至今,已经有包括bevacizumab(Avastin)、cetuximab (Erbitux)、geftinib(Irresa)、erlotinib(Tarceva)在内的8个单抗和小分子酪氨酸激酶抑制剂先后上市,超过100个药物正在进行临床研究。
最近,分子靶向抗肿瘤药物治疗又提出另一个挑战性概念:多靶标酪氨酸激酶抑制(mul-tiple targeted tyrosine kinase inhibition)的策略。基于肿瘤发生发展的复杂性,绝大部分肿瘤不是依靠某一条信号通路来维持其生长和存活的,信号通路之间存在着交叉和代偿。多靶标药物可以通过抑制多重信号通路或一条通路中上下游的多个分子而达到协同治疗、克服耐药的双重功能。这一概念已经获得了令人信服的临床证据,两个多靶点小分子化合物sunitinib和sorafenib最近已分别被FDA批准单药用于肾癌。
其中sunitinib同时靶向VEGF-2和PDGFR、KIT和FLT3等多种酪氨酸激酶。而sorafenib一方面通过抑制RAF/MEK/ERK信号传导通路直接抑制肿瘤生长;另一方面通过抑制VEGF和PDGF而阻断肿瘤新生血管的形成,间接地抑制肿瘤细胞的生长。另一个能同时抑制EGFR、VEGFR和RET酪氨酸激抑制剂Zactima(vandetanib)已获欧洲罕见病药品委员会(COMP)的批准推荐,用于髓甲状腺癌。Gleevec在临床应用中遇到的最大问题是易产生耐药性,而第二代靶向Abl和Src激酶的双重抑制剂对大部分Gleevec耐药的肿瘤都有效。可以预见,多靶点联合阻断信号传导将成为未来酪氨酸激酶抑制剂研发的新的发展方向。
二、抑制肿瘤新生血管生成(anti-angiogenesis)
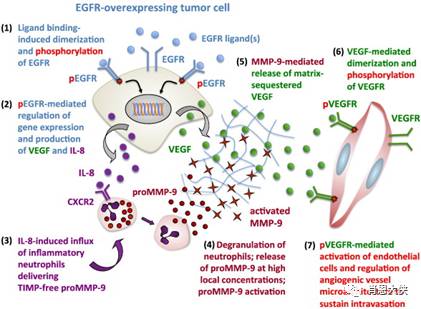
靶向VEGFR、FGFR、EGFR等具有促进肿瘤新生血管生成作用的受体酪氨酸激酶抑制剂代表了抗肿瘤靶向药物研究中另外一个重要方向——抑制肿瘤新生血管生成。自Folkman在20世纪70年代提出肿瘤新生血管生成的概念以来,靶向肿瘤新生血管生成的抑制剂研究已经取得了长足的进展,以Avastin为代表的肿瘤血管新生抑制剂得到了广泛认可,目前已有包括我国在内的28个国家批准将该类抑制剂用于肿瘤临床治疗。
这些抑制剂的研发,都是基于对肿瘤新生血管的生成过程的认识:①肿瘤组织在很长一段时间处于休眠期,依靠组织渗透维持其生长。肿瘤长到1.0~2.0 mm3时,简单的渗透作用已经不能满足生长所需要的氧气和营养物质以及代谢物的清除,瘤组织内部缺氧,缺氧诱导因子(hypoxiainducible factor,HIF)表达增强;②各类促血管生长因子如VEGF等表达上调,刺激内皮细胞活化,分泌血管生成所需的其它因子如基质金属蛋白酶(matrix metallo proteinase,MMP)等,降解基底膜和细胞外基质,内皮细胞呈游离态;③游离的内皮细胞向刺激因子迁移,开始形成血管雏形;④内皮细胞在刺激因子作用下增殖;⑤内皮细胞重新排列组合呈条索状,并刺激成纤维细胞分泌细胞外基质,形成新的血管。
针对上述过程的各个环节的抑制剂,都能不同程度的阻碍肿瘤血管的新生,减慢实体瘤组织生长速度。除了上文提到的靶向VEGF的单抗Avastin和小分子化合物sunitinib、sarafenib两代酪氨酸激酶抑制剂外,内源性的新生血管抑制剂endostatin、interferon-β、2-metho-xyestradiol、tetrahydroco-rtisol等已经分别在临床Ⅱ/Ⅲ期实验,其中endostatin已经在我国上市。另外还有MMP抑制剂Marimastat(BB-2516)、AG3340、Neovastat以及抑制内皮细胞整合素αvβ3的单抗Vitaxin和小分子抑制剂EMD121947等正在进行各期临床试验。
三、靶向细胞内的信号转导分子
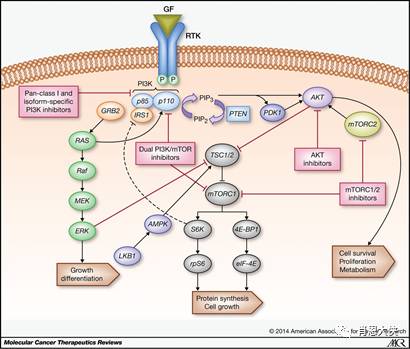
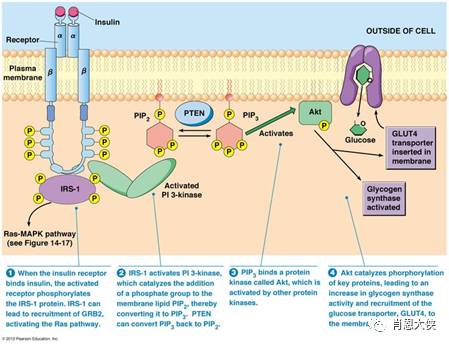
生长因子等细胞外界信号与其特异受体结合产生的刺激通过多条信号通路向细胞内传导,构成了细胞内纷繁复杂的信号转导系统,共同调控着细胞的增殖、分化。其中,由磷酯酰肌醇3-激酶(PI3K)和其下游的蛋白激酶B(PKB/Akt)、雷帕霉素靶体蛋白(mTOR)组成的PI3K-AKT-mTOR通路;丝苏氨酸蛋白激酶Ras和丝裂原活化蛋白激酶(MAPK)三级级联激酶组成的Ras-MAPK通路;以及下游信号转导与转录激活因子STAT家族与肿瘤发生、发展密切相关,已经成为抗肿瘤研究的重要靶点。
PI3K是由脂类和丝/苏氨酸激酶组成的一个庞大家族,包括数个磷酯酰肌醇激酶和DNA依赖的蛋白激酶如ATM、ATR和DNA-PK等。Akt是PI3K最主要的下游分子,前者可以激活包括mTOR在内的多个底物。mTOR分子被激活后,能通过磷酸化下游核糖蛋白S6激酶(p70S6K)和4E-结合蛋白(4E-BP)刺激细胞增殖、转化并抑制凋亡。研究发现,PI3K-AKT-mTOR在广泛的人类肿瘤谱中失调,该通路中某些基因突变所致的功能异常或缺失会引起正常细胞转化,促进肿瘤细胞增殖和存活并介导肿瘤细胞的侵袭和迁移。
因此,PI3K-AKT-mTOR信号通路已经成为一个很有希望的抗肿瘤治疗靶点。一些小分子抑制剂,如靶向PI3K的催化亚基p110的wortmannin、LY294002、IC484068和天然来源的PI3K抑制剂鱼藤素(Deguelin);抑制Akt激活所必需的丝/苏氨酸激酶PDK的straurosporine、UCN-01和Akt的抑制剂perifosine;以及特异靶向mTOR的抑制剂雷帕霉素及其类似物RAD001、CI779和AP23573等已经分别进入各期临床研究。
理论上来讲靶向PI3K-AKT-mTOR信号通路的上下游三个分子都可以有效的抑制该通路,其中靶向上游分子PI3K或Akt最有优势,一方面可以避免因抑制mTOR引起Akt的反馈性激活,另一方面可以同时阻断Akt下游的多条通路,避免不同信号通路之间的代偿作用。但是这一理论上的优势并没有在临床试验中得到证实,同时多条通路被阻断后伴随的毒性问题反而阻碍了其临床应用。而特异靶向下游mTOR分子的雷帕霉素及其衍生物在临床试验中却体现出较好的治疗效果,并显示出良好的开发前景。
Ras-MAPK通路由一组级联活化的丝/苏氨酸蛋白激酶组成,广泛存在于各种细胞中,与PI3K/Akt通路共同肩负着将膜受体信号向细胞内转导的任务,对细胞周期的运行和基因表达有重要调控作用。在多种肿瘤中都发现了该通路蛋白突变引起的持续激活,在肿瘤的发生过程中起到了重要作用。其中,Ras癌基因蛋白作为MAPK通路分子开关,可被包括EGFR、HER-2、VEGFR、PDGFR和MET等在内的多个细胞膜上的酪氨酸激酶受体激活,引发下游的级联信号通路。
在各种肿瘤中Ras的总的突变率大约为30%,是人类肿瘤中突变率最高的基因。Raf是Ras下游最重要的蛋白,通过从胞浆中转移到细胞膜上而被激活。Raf即MAPKK激酶(MAPKKK),属于丝/苏氨酸蛋白激酶,是MAPK级联反应的第一个分子,它的磷酸化启动了MAPK的三级级联激活。Ras、Raf的相继激活能活化下游的MEK、ERK等激酶,促进细胞的增殖同时还能通过磷酸化Bax、Bak抑制细胞凋亡。该通路在肿瘤发生、发展中的重要作用为抗肿瘤研究提供了多个潜在的靶点。
目前,针对上下游不同分子已经涌现出多个反义核苷酸和小分子化合物,其中最引人瞩目的有影响Ras羧基末端功能的法尼酰基转移酶抑制剂lonafarnib、tipifarnib(Zarnestra),以及上文提到sorafenib。该化合物最初被认为是Raf的抑制剂,后来发现对多个靶点包括VEGFR-2、Flt-2和c-Kit等都有作用。还有靶向下游蛋白MEK的PD0325901和ARRY-142886也已进入临床研究用于黑色素瘤病人。
STAT(Signal Transducer and Activator of Transcription)家族蛋白是一组可以被不同的生长因子受体激活的蛋白,将上游的信号传递到细胞核,通过诱导靶基因转录表达引起不同的生物效应,并保持信号在细胞内传递的内在特异性。在多种肿瘤细胞以及原位癌中都检测到了STAT家族成员的组成型激活。在STAT家族的7个成员中,目前发现STAT1、STAT3和STAT5与肿瘤的关系最为密切。STAT1对肿瘤细胞的增殖、新生血管生成起着负调控的作用,STAT1缺陷的小鼠易发生肿瘤。
与之相反,STAT3和STAT5的持续激活能上调凋亡抑制因子如Bcl-2、Bcl-xL、Mcl-1及细胞周期调控蛋白cyclinsD1/D2等基因的表达,刺激细胞增殖、抑制凋亡,被认为是该家族中最有希望的抗肿瘤作用靶点。在白血病、乳腺癌、头颈部磷癌等多种肿瘤中都发现了STAT3的组成型激活,并发现STAT3的小分子抑制剂或反义寡核苷酸可以逆转恶性肿瘤的表现型,增加耐药的肿瘤对化学药物的敏感性。目前靶向STAT家族的反义核苷酸和小分子抑制剂的抗肿瘤药物研发已经引起了广泛关注。
四、靶向细胞周期蛋白
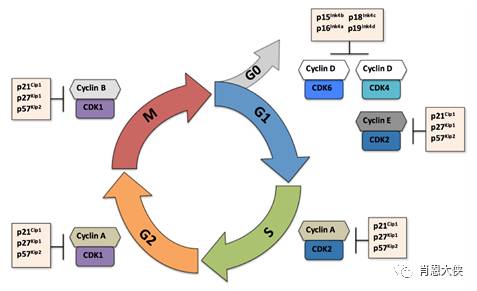
细胞生长分裂必须依次经过准备阶段的间期(interphase)和有丝分裂期(mitosis)。间期(包括G1、S、G2期)的各项生命活动保证了M期分裂时所需的细胞内各成分的复制,每次有丝分裂的结束到下一次有丝分裂的结束构成一个完整的细胞周期。细胞周期的运行与否,受控于精密的细胞周期调控机制。该调控系统的核心是一组细胞周期依赖性蛋白激酶(Cyclin-dependent-kinase,CDKs),它们各自在细胞周期内特定时间被激活,通过磷酸化对应的底物,驱使细胞周期的完成。CDKs的时相性激活依赖于时相表达的周期素(cyclin)以及周期素依赖性激酶抑制剂(cyclin-dependent kinase inhibitors,CKI)控制。
另外,除了这种正常生理条件下的周期进程调控,在长期的进化过程中,细胞建立了一套保证细胞周期中遗传信息的完整性和准确性的检查机制,即细胞周期检查点(checkpoint)。当细胞周期进程中出现异常事件,如DNA损伤或DNA复制受阻时,这类调节机制就被激活,及时中断细胞周期的运行。待DNA修复或排除了故障后,细胞周期才能恢复运转。
在细胞的癌变过程中,通常都伴随着cyclin的过度表达和CKIs的缺失,CDK的活性失去控制,细胞周期处于失控状态;肿瘤细胞的另外一个特点是细胞周期检查点缺陷,造成对细胞损伤应答的缺失。然而,这种周期检查点关卡的缺失,使得细胞对外界的损伤更加敏感又能被应用于肿瘤的治疗,增加放化疗的敏感性。基于肿瘤细胞的上述特点,恢复肿瘤细胞的周期调控和取消检查点等都成为潜在的抗肿瘤作用靶点。具体策略包括对CDK的直接催化抑制,阻碍CDK的激活,干扰周期素与CDK的相互作用,影响周期素水解失活和抑制细胞周期检测点等。
目前,已经有多个细胞周期的调节剂进入了临床研究,其中植物来源的黄酮类物质flavo-piridol能明显抑制CDK1、CDK2和CDK4,阻碍细胞通过G1/S和G2/M期检测点,能抑制多种肿瘤细胞的生长,已经处于临床Ⅱ期研究。另外,星型苞菌素(Stauros-porine)的类似物UCN-01除了抑制PKC外,还可直接抑制CDK1和CDK2的活性和细胞周期检测点激酶chk1的活化,目前正在美国和日本进行临床Ⅱ期实验。还有Paullones类似物、嘌呤霉素类似物(Pruines)等都对不同的CDK分子显示出抑制作用。
五、组蛋白去乙酰酶抑制剂(histone deacetylase inhibitor,HDACi)
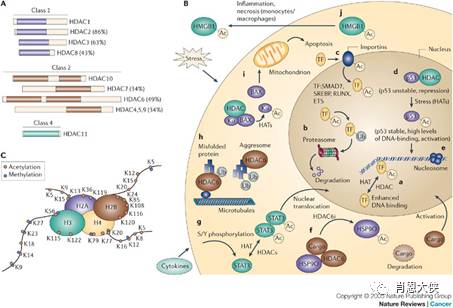
肿瘤的发生和诸多基因特别是癌基因的异常表达密切相关,而染色体结构是调控基因表达的重要因素。通常情况下,凝缩的染色体会抑制基因的转录,而有转录活性的基因一般位于松散的染色体区域。染色体的基本单位核小体是由组蛋白(Histone)和DNA组成的,其中组蛋白的转录后修饰,包括乙酰化、磷酸化和甲基化能够改变核小体的高级结构,进而影响着染色体的高级结构和基因的转录调控。细胞内一对功能相互拮抗的蛋白酶,组蛋白乙酰基转移酶(Histone acetyl-transferases,HATs)和组蛋白去乙酰酶(Histone deace-tylases,HDACs)共同决定着组蛋白的乙酰化和去乙酰化。HAT可乙酰化组蛋白末端碱性氨基酸的氨基,使核小体舒展,激活基因转录。而HDAC与之功能相反,抑制基因转录。
近些年的研究发现HDAC作为调控基因转录的关键蛋白酶,其功能异常与肿瘤的发生和发展有直接关系。当HDAC过度表达并被转录因子募集时,会抑制某些基因的正常表达。这种因HDAC活性过高引起的异常转录抑制在肿瘤中非常普遍,因此HDAC成为抗肿瘤药物最具潜力的靶点之一。抑制HDAC的活性能引起组蛋白高度乙酰化,重新激活某些抑癌基因的转录并引起多项下游效应,包括促进肿瘤细胞分化、使肿瘤细胞阻滞于G1或G2期以及诱导肿瘤细胞凋亡,从而实现其抗肿瘤作用。另外,研究发现HDACi还能激活主要组织相容性复合物、细胞间粘附分子ICAM-1、干扰素Ⅰ/Ⅱ等分子的转录,促进免疫细胞的识别和激活。HDACi还能抑制缺氧诱导的VEGF表达,抑制新生血管生成。
来自真菌的TrichostatinA(TSA)是发现的首个能高效抑制HDAC的羟胺类天然产物,但存在着天然含量低、体内代谢不稳定的缺点。目前已经有10多个不同结构类型的HDACi进入了Ⅰ/Ⅱ期临床试验,用于白血病和实体瘤的治疗。这些药物大多能在有效剂量显示出较好的耐受性,并显示出抗p-糖蛋白介导的多药耐药作用。
六、靶向泛素-蛋白酶体通路(ubiquitin-proteasome system, UPS)
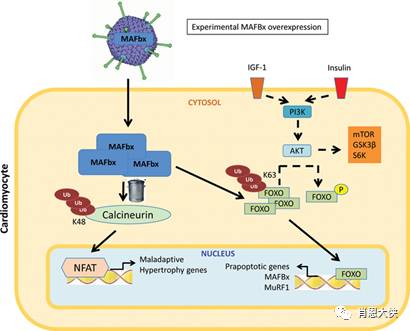
蛋白降解调控是细胞信号转导的一个重要方面,与基因转录水平的调控相比,这种转录后调控还能保证细胞在遇到外界刺激时更加快速的做出反应。UPS是真核细胞内依赖ATP的非溶酶体蛋白质降解途径,负责调控细胞内多种蛋白的水解过程,其中包括许多调节细胞生长、信号转导、基因转录和凋亡的重要分子。泛素介导的蛋白降解是一个复杂的多级反应,其过程主要是利用泛素活化酶E1、泛素结合酶E2与泛素-蛋白连接酶E3,将泛素连接至目标蛋白作为标识,并送至20S蛋白酶体进行降解,最后由泛素分解酶将泛素分解并回收再利用。
由于UPS通路与肿瘤的发生、生长和转移都密切相关,该级联反应中各个环节都成为抗肿瘤药物作用的潜在靶点。例如,通过阻断泛素分子C末端的腺苷酸化或与ATP分子竞争结合的策略来阻碍泛素的激活;根据E1与E2相互作用的结合域特异地设计能够干扰其相互作用的小分子化合物阻碍泛素分子在E1和E2之间的传递等。其中特别值得一提的是靶向E3连接酶和其下游的蛋白酶体。
E1、E2和E3构成了金字塔形的级联放大系统,最下游的E3通过识别不同底物决定着整个泛素化过程的特异性。抑制泛素连接酶E3的功能可以通过抑制其与上游E2或下游蛋白底物的作用两条途径来实现。目前还没有发现针对E3和E2相互作用的抑制剂,但是对后者的研究已经取得了一些进展,其中最典型的例子就是MDM2对p53蛋白降解的调控。MDM2分子具有E3连接酶活性,通过泛素化调控p53的降解。
Nutlins是首个发现的阻碍p53和MDM2相互作用的小分子抑制剂,其空间构象和p53分子中与MDM2作用的氨基酸残基非常相似,可以与p53分子竞争MDM2的结合位点。Nutlins在体内外都体现出抗肿瘤活性,对正常组织没有明显毒性。另外一个小分子抑制剂RITA早在1990年就发现其抗肿瘤活性,但在最近才发现其作用机理是与p53的N端结合,阻碍了MDM2对p53的识别,并稳定p53分子N端α-螺旋结构域。
E3连接酶下游的多个环节,包括蛋白酶体、参与泛素游离再循环的金属异肽酶(metal-loisopeptidase)以及对多聚泛素链的识别等都可能影响UPS通路。其中,首个上市的以UPS为靶点的小分子抑制剂bortezomib(Velcade,PS-341)就是直接抑制于蛋白酶体活性。该化合物已先后于2003年5月和2004年4月被美国FDA和欧盟药品审评管理局(European Agency for the Evaluation of Medicinal Products ,EMEA)批准用于复发性和难治性多发性骨髓瘤的治疗。最近FDA又批准其作为一线药物用于已接受过至少一次治疗的多发性骨髓瘤患者。进一步在血液系统肿瘤、实体瘤以及非霍奇金淋巴瘤中的应用正在研究中。
七、靶向DNA损伤修复系统
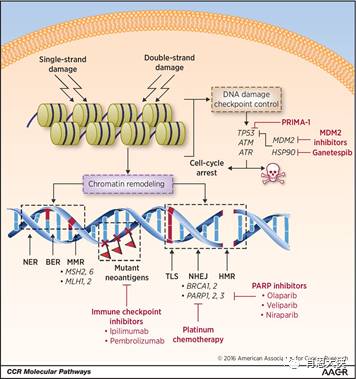
除了上述直接影响肿瘤细胞增殖调控的信号通路外,特异靶向DNA损伤修复通路中的一些关键分子也成为抗肿瘤药物研发的一个重要方向。很多传统的抗肿瘤药物包括烷化剂、DNA嵌入剂、拓扑异构酶抑制剂、抗代谢物等都是通过直接或间接造成不同形式的DNA损伤来实现其抗肿瘤作用。
在外界损伤的刺激下,细胞能启动6条修复通路来分别应对不同类型的损伤:①直接修复(direct repair,DR)通路修复O6-烷基鸟嘌啉引起的损伤;②碱基切除修复(base excision repair,BER)针对因氧化还原或烷基化引起的碱基损伤;③核苷酸切除修复(nucleotide excision repair,NER)修复因辐射、化学药物或蛋白-DNA交联引起的核苷酸水平的损伤;④碱基错配修复(mismatch repair,MMR)纠正碱基错配;⑤同源重组修复(homologous repair,HR);⑥非同源的末端连接(non-homologous end-joining,NHEJ)通路,其中后两条通路专门修复DNA双链断裂(DNA double strand breaks,DSBs)。这些通路的激活往往削弱了化疗药的抗肿瘤效果,成为产生耐药的一个重要因素。
八、靶向肿瘤基质细胞(微环境)药物
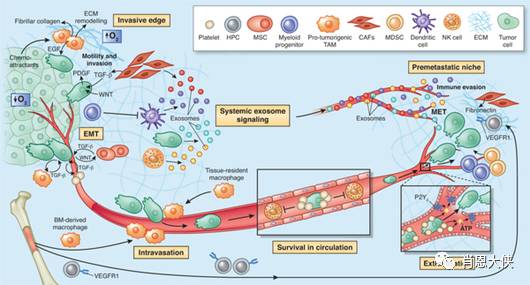
1 成纤维细胞
成纤维细胞是最主要的基质细胞,癌相关成纤维细胞(CAFs)也被称为活化的成纤维细胞或肌性成纤维细胞,其所分泌的基质衍生因子-1(SDF-1,又称CXCL12)可直接刺激CXCR4+肿瘤细胞生长,还可募集CXCR4+内皮前体细胞(EPCs)参与肿瘤的血管生成。活化的纤维细胞通过SDF-1/CXCR4趋化轴吸引CXCR4+肿瘤细胞做定向迁移。
肿瘤原发灶所分泌的生长因子刺激“预转移灶”内成纤维细胞、血小板衍生的生长因子受体阳性(PDGFR+)细胞和纤维连接蛋白增多,为肿瘤细胞增殖提供环境。在微转移灶的缺氧环境中,活化的成纤维细胞可产生血管内皮生长因子A (VEGF-A)以促成血管生成,募集来的造血祖细胞(HPCs)也促进血管的生成。
肿瘤细胞与局部或远处成纤维细胞之间通过旁分泌或内分泌而发生交互作用,肿瘤细胞通过这种机制调节肿瘤微环境并使远处组织发生显著改变。癌细胞分泌的白细胞介素-1(IL-1)、成纤维细胞生长因子-2(FGF-2)和PDGF诱导成纤维细胞分泌肝细胞生长因子(HGF),HGF与癌细胞上的c-Met分子结合后能增强癌细胞的侵袭和迁移能力。另外,转化生长因子-β(TGF-β)、表皮生长因子(EGF)、胰岛素生长因子(IGF)和Wnt1等,是实现瘤细胞和基质细胞之间“对话”(cross-talk)的旁分泌信使。
2 浸润的炎症/免疫细胞
肿瘤浸润的炎症细胞是一把双刃剑,除有一定的抗肿瘤作用外,更多情况下是在促进肿瘤的发生和发展。在众多浸润的炎症细胞中,以肿瘤相关巨噬细胞(TAMs)的研究最为深入广泛。TAMs可通过分泌 FGF、HGF、EGF、PDGF和TGF-β等多种生长因子促进肿瘤生长。
单核细胞是TAMs的前体细胞,肿瘤产生的CCL2/MCP-1吸引单核细胞到肿瘤部位并分化为TAMs。肿瘤细胞、成纤维细胞、内皮细胞和TAMs都能通过产生CCL2、CCL5、CXCL8/IL-8和SDF-1进一步募集单核细胞。另外,CSF-1、VEGF-A和胎盘生长因子(PIGF)等也能引发单核细胞向肿瘤组织浸润。缺氧介导的缺氧诱导因子-1(HIF-1)和VEGF也能吸引TAMs向肿瘤缺氧区集聚。纤溶酶原片段K1-3能阻断TAMs的迁移和肿瘤对其募集作用。
TAMs不仅直接或间接地释放血管生成因子来促进血管芽生,而且能够产生一些酶来参与血管的重建。TAMs是VEGF-A的重要来源之一,还可通过分泌MMP来释放细胞外基质(ECM)中被结合的VEGF-A。TAMs受缺氧和CSF-1等因素调节,缺氧使HIF-1和HIF-2调节的启动子发生转录性活化,上调VEGF-A、MMPs、白介素和趋化因子。TAMs在破坏基底膜、引发癌细胞的迁移方面也发挥着重要作用。TAMs所分泌的MMP2、MMP9、TGF-β、uPA、tPA和组织蛋白酶等降解胶原、层粘连蛋白和纤维连接蛋白等ECM成分,进而促进肿瘤的侵袭和转移。
3 未分化的骨髓细胞
在肿瘤生长的早期,VEGF-A和其他细胞因子能把骨髓中的内皮祖细胞(EPCs)动员到外周血,使之成为循环的内皮前体细胞(CEPs)并最终整合到新生血管的管壁上。肿瘤所分泌的生长因子和趋化因子会引起骨髓细胞增殖和向肿瘤内聚集。肿瘤细胞分泌的VEGF-A和PIGF等能把VEGFR-1+的HPCs和VEGFR-2+的EPCs募集到肿瘤的新生血管部位,促进肿瘤的生长和血管生成。
来自骨髓的造血祖细胞和未成熟的髓系细胞在SDF-1/CXCR4和CXCL5/CXCR2生物轴作用下被募集到肿瘤侵袭前沿,通过分泌金属蛋白酶来增强肿瘤的外侵和转移,并能促进肿瘤血管和淋巴管生成。CD11b+Gr-1+髓系抑制细胞(MDSCs)通过分泌抑制免疫反应的细胞因子、上调NO、产生活性氧族以及增强L精氨酸酶的活性而抑制免疫反应,引起肿瘤的免疫逃逸。
近来的研究发现,造血祖细胞能为肿瘤细胞在远处的植入和增殖做好准备,原发灶所释放的特殊趋化因子能动员一些未成熟的骨髓来源细胞(BMDCs)成群地植入到将要发生转移的远处靶器官内(图1-1B),分泌MMP-9等降解基质,使周围环境更适合肿瘤的种植和生长。另外,BMDCs也表达CXCR4,通过与CAFs相互作用而增加SDF-1生成,进而吸引CXCR4+肿瘤细胞。
4 内皮细胞、周细胞和血小板
血管内皮细胞迁移、血管出芽是血管生成的主要模式。周细胞在PDGF-B作用下被募集到新生血管周围,通过加强血管外侧的细胞间紧密连接以维持血管的稳定性。血小板所提供的信号能够引导BMDCs和瘤细胞的归巢与滞留,血小板所释放的SDF-1在募集和“挽留”CXCR4+的HPCs和EPCs方面起到关键作用,并趋化CXCR4+肿瘤细胞。穿梭在原发灶、转移灶和骨髓之间的血小板不断释放大量的细胞因子,从而把这些部位连接在一起。
5 趋化因子及其受体与肿瘤的生长和转移
趋化因子是指在多种炎症和非炎症状态下调节白细胞和其他一些类型细胞进行流动和活化并对这些细胞具有定向趋化作用的细胞因子。目前发现大约50个趋化因子和20个趋化因子受体,根据N末端两个半胱氨酸的位置,趋化因子被分为4类:CXC, CC, CX3C 和 C。
在肿瘤的缺氧环境中,成纤维细胞分泌的CXCL12(SDF-1)和肿瘤细胞表达的CXCR4都增加,从而刺激肿瘤细胞移动和侵犯。另外,多种趋化因子有促进肿瘤血管生成的作用,如CXCL1,CXCL2, CXCL3, CXCL5, CXCL6,CXCL7, CXCL8等。CXCR2不仅在肿瘤血管形成中扮演重要角色,而且能向肿瘤微环境中招募巨噬细胞。
肿瘤细胞通过趋化因子来募集内皮细胞、颠覆免疫监视、操纵免疫细胞(免疫编辑)并最终导致免疫逃逸,从而促使肿瘤生长和向远处转移。肿瘤细胞不仅分泌趋化因子,也能对趋化因子作出反应。肿瘤转移的靶器官能分泌的大量趋化因子,由于肿瘤细胞表达一些趋化因子的受体,在趋化因子轴的作用下,引导肿瘤细胞向靶器官转移(肿瘤转移的“信号或归巢”signaling or homing hypothesis)。CCR7/CCL21轴介导肿瘤细胞向前哨淋巴结转移,CCR7/CCL21轴介导肿瘤细胞向相关淋巴结转移,而CCR10/CCL27生物轴则参与调节黑色素瘤的皮肤转移。
6 以肿瘤微环境为靶点的治疗策略
理论上直接作用于肿瘤细胞的治疗方法有许多不足之处,如肿瘤之间和肿瘤组织内部肿瘤细胞之间的异质性,是造成疗效差异的主要原因;肿瘤细胞生物或遗传特点的不稳定性,在疾病进展和治疗过程中,这种不稳定性会不断增加;目前以肿瘤细胞为目标的治疗措施的疗效仍有限;在以肿瘤细胞为靶点的治疗过程中常有耐药的 肿瘤细胞克隆出现。
而靶向于肿瘤微环境的治疗策略有其自身的的优势,如肿瘤间质细胞具有稳定的遗传背景,不易出现突变和耐药发生;肿瘤微环境的异质性更小,疗效相对稳定,并有可能预测肿瘤组织对治疗的反应性;在控制肿瘤转移方面可以发挥极为重要的作用;化疗和放疗等多种治疗措施对间质的作用也是发挥疗效的重要侧面,有时也可 能是最终的作用路径。
7 靶向作用于肿瘤微环境的药物
以肿瘤微环境中的成分作为新靶点的治疗方法能克服许多目前传统治疗方式的限制。针对肿瘤基质的靶向治疗,由于目标的特异性,将不会有太大的毒副作用。不同于肿瘤细胞,内皮细胞的基因稳定,因此不太可能获得耐药性突变。此外,针对肿瘤微环境中多个成分的靶向治疗的联合应用,可以避免单一靶向时肿瘤通过代偿弥 补肿瘤生存所需环境物质。此外,联合方案可降低各自的剂量,从而减少治疗相关的毒副作用。此疗法的最终目标提高治疗效果、延长患者生存。
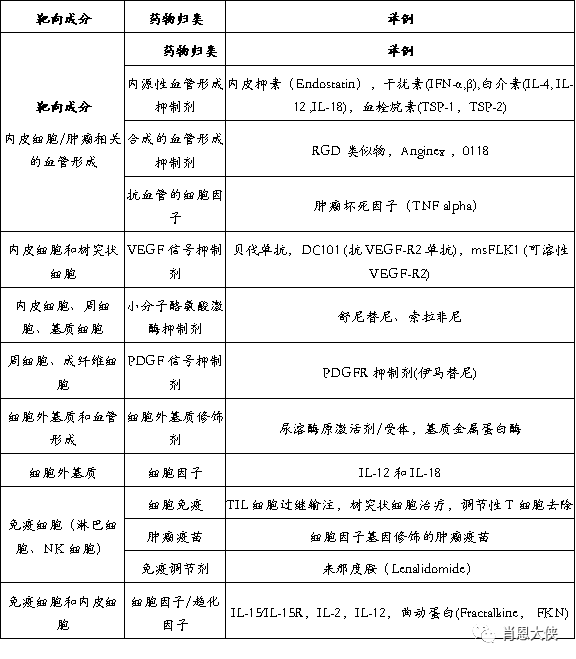
RGD:精氨酸-甘氨酸-天冬氨酸(arginine-glycine-aspartic acid);TNF:肿瘤坏死因子;VEGF:血管内皮生长因子;PDGF:血小板衍生的生长因子;ECM:细胞外基质
九、靶向肿瘤干细胞的治疗策略
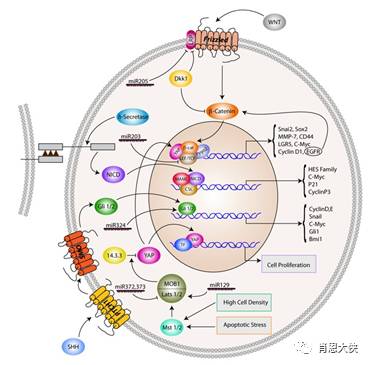
肿瘤干细胞(cancer stem cell/tumor-initiating cell, CSC/TIC )是肿瘤细胞中具有干细胞特性的细胞亚群,其增殖能力明显强于同一肿瘤组织中的其他癌细胞, 在肿瘤的发生、发展和维持中起十分重要的作用,并且在动物体内表现出很强的成瘤能力。Lapidot等人于1994年报道急性髓系白血病(acute myeloid leukemia, AML)中存在表型为CD34+CD38-的细胞亚群, 将其注入非肥胖糖尿病/严重联合免疫缺陷型(nonobese diabetic/severe combined immune-deficient, NOD/SCID)小鼠体内可诱发白血病。
Muhammad等人在2003年发现乳腺癌中也含有能够在NOD/SCID小鼠中连续成瘤的干细胞样的癌细胞群体,这是首例关于肿瘤干细胞存在于实体瘤中的报道。至目前为止, 人们相继在多种原发性肿瘤和癌细胞系中鉴定到肿瘤干细胞的存在。已发现有肿瘤干细胞存在的肿瘤种类。肿瘤干细胞与增殖力较弱的非干细胞样肿瘤细胞相比表现出更强的形成肿瘤的能力。O'Brien等人从结肠癌中筛选到含有肿瘤干细胞的CD133+细胞亚群, 在NOD/SCID小鼠成瘤实验中显示出与CD133-细胞亚群在成瘤能力上的明显差异。1×103个CD133+细胞足以在小鼠体内形成肿瘤, 且成瘤率高达100%, 而CD133-细胞的注射量达到2.5×105时, 仅得到11%的成瘤小鼠。
此外,与肿瘤细胞相比, 肿瘤干细胞在放化疗中表现出更强的抗性, 这与肿瘤干细胞能更有效的启动DNA应急反应和高表达耐药相关蛋白有关。肿瘤干细胞的高成瘤率和对放化疗的抵抗能力,使其在放化疗过程中得以存活, 导致肿瘤的复发。因此, 有效的清除肿瘤干细胞对于获得理想的抗癌疗效十分必要。
各种肿瘤类型中肿瘤干细胞的分子标记(或分选方法)
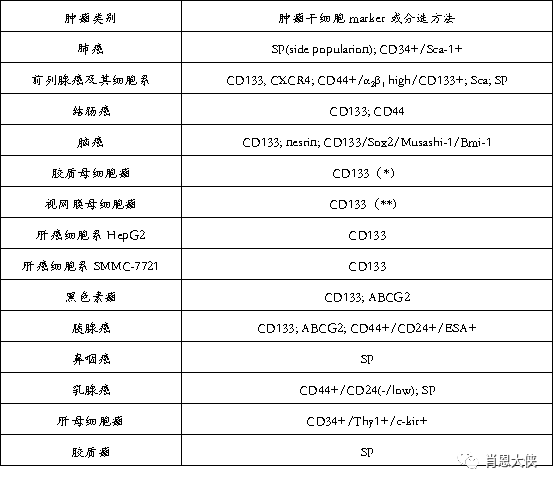
(*) 少数样品肿瘤干细胞为CD133阴性, (**)待证实
载体直接影响治疗作用的发挥, 因此选择合适的载体对于取得理想的疗效很关键。目前, 在靶向肿瘤干细胞的研究中应用最多的载体是小分子抑制剂和融合蛋白。然而,由于靶向性差, 易产生副作用等缺陷, 限制了此类药剂在临床治疗中的应用。大量研究表明, 病毒尤其是溶瘤腺病毒(oncolyticvirus)比小分子类和蛋白类药物更适合作为靶向肿瘤干细胞的载体。
以溶瘤腺病毒为载体的病毒基因治疗(gene-virotherapy)方案已在实体瘤治疗中取得巨大的成功, 将其应用于靶向肿瘤干细胞的治疗可望获得更好的疗效。此外,肿瘤干细胞的特性和内部的分子机制与正常干细胞很相似。根据此特点, 人们制定了一系列具有针对性的治疗策略,如抑制增殖, 促进分化, 诱导凋亡,破坏微环境(niche)和增强放化疗敏感性等。初步实验结果表明,这些策略能有效的靶向肿瘤干细胞并抑制其功能, 达到理想的治疗效果。以下将对靶向肿瘤干细胞治疗载体的选择以及具体靶向策略等问题作一介绍。
1 靶向肿瘤干细胞的载体
目前应用于肿瘤干细胞靶向性治疗的载体可分为小分子化学物质, 蛋白类制剂和病毒。其中, 溶瘤腺病毒同时具有介导外源基因体内表达和直接裂解肿瘤细胞两种治疗功能,在各种载体中显现出独有的优势。
2 小分子和蛋白类药物
肿瘤干细胞靶向性治疗的研究当前主要集中于对特效性化学小分子类和蛋白类治疗药物的筛选。其中, 小分子药物具有易于制备, 适合产业化和无免疫原性等优点, 因而在靶向肿瘤干细胞尤其是白血病干细胞的研究中涉及较多。Guzman等人发现蛋白酶体抑制剂MG-132和蒽环类抗生素idarubicin可以选择性作用于AML干细胞, 而对正常造血干细胞没有影响。
后来, 该研究小组又筛选出一种倍半萜烯内酯Parthenolide药物, 能够诱导粒细胞性白血病(myelogenous leukemia)干细胞发生凋亡。最近有报道证实,小分子抑制剂RHPS4和砷均能够抑制白血病干细胞的增殖能力。但是小分子药物副作用明显,靶向性差, 多次给药可引发耐药性, 这些缺陷大大限制了该类制剂在实际治疗中的作用。
将抗体和毒素融合制成的嵌合体-免疫毒素(immunotoxin),可以将起杀伤作用的多肽或蛋白导入表达相应抗原的靶细胞内, 对其实行选择性攻击,而不影响其他细胞。已知肿瘤干细胞存在于有特定表面抗原表型的细胞亚群中, 这为针对性治疗的实施提供了有效靶点。部分肿瘤干细胞的抗原表型。
目前,已有研究小组研制出对肿瘤干细胞具有靶向性的immunotoxin。例如,Du等人将CD123抗体与假单胞菌外毒素A重组构成immunotoxin, 体外实验表明该融合蛋白能有效的靶向AML白血病干细胞。Feuring-Buske等人利用IL-3与白喉毒素的融合蛋白杀灭AML干细胞, 效果显著且不会影响正常造血干细胞。虽然含有特异性抗体的融合蛋白与小分子相比,靶向性得到很大提高, 但免疫原性强和生产成本高仍是其需要克服的缺点。
3 病毒载体
与使用小分子类和蛋白类药物的治疗方案相比, 以病毒为载体的基因疗法(gene therapy)则表现出许多明显的优势:(ⅰ)能够介导外源基因高效的进入细胞并长期表达;(ⅱ)某些种类的病毒对肿瘤细胞有天然的靶向性, 经改造后, 靶向性和安全性显著提高;(ⅲ)不易使机体产生耐药性。Clement等人使用慢病毒为载体介导的RNA干扰(RNAi), 显著抑制了胶质瘤细胞的SHH通路活性, 削弱了CD133+细胞在NOD/SCID小鼠体内的成瘤能力, 有效的靶向了肿瘤干细胞群体。其中, 慢病毒能维持治疗基因长期表达的优势在实验中得到体现。
目前, 用于肿瘤基因治疗的病毒类载体类型很多。其中, 逆转录病毒最早被用于基因治疗,其特点是仅整合分裂期细胞。Ho等人在检测MCM的mRNA表达水平时发现大部分的肿瘤干细胞处于G0期。而且也另有文献指出肿瘤干细胞分裂不活跃。所以, 尽管逆转录病毒对肿瘤中分裂旺盛的细胞特异性很强,但并不是靶向肿瘤干细胞的理想载体。慢病毒克服了逆转录病毒对静止期细胞不能整合的缺点, 还能够介导外源基因长期表达。但是其产量过低, 即便是第一代慢病毒的产量也不过108-109pfu/mL, 而现在应用最广的第三代慢病毒的滴度只有106pfu/mL, 不能满足实际治疗对病毒量的需求。
腺相关病毒虽然具有易于操作, 安全性较好,组织特异性靶向等优点, 但其可插入片段的最大长度只有4.7kb,不能同时携带多个基因, 所以其在靶向肿瘤干细胞治疗中的应用受到一定限制。痘病毒和单纯疱疹病毒也分别由于免疫原性强和可转染细胞的种类有限而不适合作为靶向肿瘤干细胞的载体。
改造后的腺病毒是使用非常广泛的一种肿瘤治疗载体, 其相关研究比较深入, 可插入的基因片断容量大(上限至38kb), 可感染的细胞类型多, 转染效果好, 包装滴度非常高(1011-1012pfu/mL), 能够同时转染分裂期和非分裂期的细胞, 安全性较好,同其他类型的病毒载体相比, 更适宜成为靶向肿瘤干细胞的载体。不过,包括腺病毒在内的病毒类载体大都为复制缺陷型, 其复制和感染过程中的关键基因已被敲除或突变。经过这种改造的病毒不能在细胞内复制,仅能发挥介导基因在细胞内表达的载体功能, 治疗潜能的发挥受到很大限制。
4 新型靶向肿瘤干细胞载体-溶瘤腺病毒
除了通过介导治疗基因的表达来抑制肿瘤的发展外, 病毒还能够通过自身的复制直接裂解肿瘤细胞。1997年, Heise等人改造了腺病毒的E1B区, 使55kD蛋白缺失, 使之选择性的在p53异常的肿瘤细胞中增殖,治疗效果让人振奋, 这就是世界上第一个构建成功的溶瘤腺病毒。
实验室的溶瘤腺病毒载体ZD55也是基于上述原理构建的。该类复制型腺病毒能够在肿瘤细胞内大量增殖, 最终致使癌细胞裂解而引起瘤体的消退。另外,溶瘤病毒还可以通过诱发机体的免疫反应来增强免疫系统的局部抗肿瘤作用。为了避免溶瘤腺病毒对正常组织产生毒副作用,使其复制能力局限于肿瘤细胞, 人们进行了大量的改造工作, 采用修改病毒自身基因或使用肿瘤特异性启动子调控病毒复制相关基因表达的策略构建了多种能安全性较高的肿瘤特异性溶瘤腺病毒。
最近, Jiang等人通过实验发现, 溶瘤腺病毒Delta-24-RGD能够有效的清除脑癌干细胞。这是首例以溶瘤腺病毒为治疗载体成功杀伤肿瘤干细胞的报道, 表明了将其应用于靶向肿瘤干细胞治疗的有效性。更重要的是,肿瘤干细胞中人端粒酶(hTERT)的活性明显高于正常干细胞, 而我们自行构建的溶瘤腺病毒载体CHNK300和Ad-TERT使用hTERT启动子调控病毒复制中起关键作用的E1A蛋白, 能够选择性的在hTERT转录活跃的肿瘤干细胞中复制而不影响正常干细胞,在杀伤肿瘤干细胞的同时又能有效降低毒副作用的产生, 作为靶向肿瘤干细胞的载体非常理想。
病毒基因治疗策略结合了基因疗法和病毒疗法两种治疗方案的优点, 使用溶瘤腺病毒为治疗载体, 介导外源基因进入癌细胞并使之有效表达。该策略不仅可以依靠增殖型病毒的溶瘤作用来直接杀伤癌细胞,还能使携带的治疗基因拷贝数随病毒复制而大量扩增, 显著提升外源基因的治疗效果。将构建的携带鼠源内皮抑制素(mouse endostatin, mE)的溶瘤腺病毒CHNK300注射到小鼠体内诱发的肿瘤中,28天后, 肿瘤体积消减至770mm3, 而注射了携载mE的复制缺陷性腺病毒的对照组瘤体仅减小至1400mm3。
大量的证据表明, 以溶瘤腺病毒为载体的病毒基因治疗策略表现出良好的应用前景,其治疗的效果明显好于单独采用溶瘤策略或传统基因治疗策略所达到的疗效, 是最有发展前途的肿瘤基因治疗方案之一。若使溶瘤腺病毒携带针对肿瘤干细胞的治疗基因,应用于靶向肿瘤干细胞的治疗, 可望取得令人满意的疗效。
5 针对肿瘤干细胞的治疗策略
为了限制肿瘤干细胞在肿瘤发生和维持中的功能, 取得更为理想的治疗效果, 研究人员采取了多种行之有效的针对性策略。如抑制增殖,促进分化, 诱导凋亡, 破坏niche和增强放化疗敏感性等。
(1)削弱增殖能力
WNT, SHH和Notch等细胞信号通路以及人端粒酶(hTERT)不仅调控干细胞的自我增殖, 而且在生物体的发育, 成体后组织的恒态维持(homeostasis)中起关键作用, 其异常可导致肿瘤的发生。最近的研究表明, 肿瘤干细胞功能的维持同样依赖这些通路。因此, 抑制上述信号通路和hTERT的活性将对肿瘤干细胞自身功能的维持产生很大影响, 从而削弱其增殖能力。
Fan等人用阻碍Notch通路的γ分泌酶抑制剂处理成神经管细胞瘤, 明显削弱了CD133+肿瘤细胞的增殖力, 但对CD133-的肿瘤细胞分裂没有影响。Verma等人利用RNAi技术, 下调结肠癌WNT通路中关键蛋白β-catenin的表达, 数据显示, 被处理的细胞在软琼脂上形成克隆的比率和在裸鼠体内成瘤的能力明显下降,表明肿瘤干细胞的增殖能力被有效抑制。Phatak等人使用小分子药物RHPS4选择性抑制白血病干细胞的端粒酶催化亚基(hTERC), 减弱了其在核内的活性,结果造成白血病干细胞增殖能力明显下降。
另外, 胚胎干细胞增殖和全能性维持所需的Oct3/4、Nanog和Sox2因子等, 对肿瘤干细胞发挥功能也起到重要作用,提示这些因子也可以作为肿瘤干细胞的靶点, 用以削弱其增殖能力。
(2)促进分化
肿瘤可以视为异常发育的器官。与正常器官发育的不同之处在于, 肿瘤组织中的干细胞往往分化异常, 表现为分化过程受阻。因此, 诱导肿瘤干细胞重新分化, 使其丧失分裂潜能, 同样可以到达抑制肿瘤的发展的目的。
全反式维甲酸(all-transretinoic, ATRA)最早被用于肿瘤的促分化治疗, 在对急性早幼粒细胞性白血病(Acute promyelocytic leukemia, APL)的治疗中表现出良好的疗效。Bergmann等人使用组蛋白去乙酰化酶的抑制剂SAHA处理乳腺癌细胞MCF-7, 可观察到明显的细胞分化特征。另外, 化疗药物Cannabinoid也被证实具有诱导胶质瘤干细胞分化和抑制肿瘤发展的作用。骨形态发生蛋白(Bone morphogen protein, BMP)是一类在多种组织发育中起作用的细胞因子。其中BMP4对神经组织的发育和神经干细胞的正常有序分化发挥重要功能。最近,Piccirillo等人用外源的BMP蛋白诱导胶质母细胞瘤干细胞发生分化,结果导致整个肿瘤群体的成瘤能力被严重削弱。
(3)破坏微环境
成体组织中的干细胞均处于niche中。Niche为干细胞提供养分和保护, 使之免受外来物质的毒害,协助其维持自我增殖能力, 调控其分化进程。因此, 破坏niche会严重影响干细胞的正常功能, 甚至会导致死亡。已有证据证明肿瘤干细胞也处于niche中以维持其自我更新能力和特性。破坏肿瘤干细胞所依赖的niche,会严重影响肿瘤干细胞数量和功能的维持。
Calabrese等人证实脑癌的肿瘤干细胞位于内皮细胞附近的区域,脱离该区域会影响肿瘤干细胞的增殖能力。此外, 在小鼠脑内成瘤实验中,与人内皮细胞共注射的脑癌干细胞的增殖能力明显强于单独注射的脑癌干细胞, 表明破坏肿瘤干细胞niche会严重影响其功能的维持。AML干细胞为维持其增殖能力必须进入位于骨髓的niche中, 细胞表面的跨膜蛋白CD44, 在其向骨髓迁移的过程中起重要作用。Jin等人用CD44的单克隆抗体H90阻断CD44与其功能受体的结合, 以阻碍AML干细胞向niche转移, 结果大大抑制了其增殖和成瘤的能力。
(4)诱导凋亡
目前, 通过诱导凋亡程序的启动消灭肿瘤细胞的策略已经被广泛采纳。该策略可以通过两种途径实现: 促进促凋亡通路的活性和抑制抗凋亡通路的功能。研究表明,参与这两种通路的蛋白种类繁多。其中较为常见的促凋亡蛋白包括: p53, TRAIL, caspase3,GranzymeA/B, Bid, Bax和Fas等; 重要的抗凋亡蛋白包括: Bcl-2, Bcl-xL, survivin和XIAP等。通过载体介导外源促凋亡蛋白的表达, 或使用RNAi技术抑制抗调亡蛋白的翻译, 是目前常用的诱导细胞凋亡的手段。
Zhang等人使用溶瘤腺病毒ONYX-411携带能沉默突变型K-ras的siRNA转染特定癌细胞,能有效促进肿瘤细胞凋亡的发生。如果更进一步, 不是仅仅通过单个途径,而是在表达促凋亡蛋白的同时抑制抗凋亡通路的活性, 则能更有效的诱导癌细胞凋亡。Pei等人使用溶瘤腺病毒ZD55介导Smac和TRAIL在肝癌细胞内同时表达, 在抑制抗凋亡蛋白表达的同时促进凋亡通路的活性, 与单独使用Smac或TRAIL相比,能更好的杀死肿瘤细胞。
Lebedeva等人将白介素-24(IL-24)和一段能沉默突变型K-ras基因的反义RNA(K-ras AS)同时插入复制缺陷型腺病毒中构建成携带双基因的载体, 经实验证明比只携带IL-24或K-ras AS的腺病毒有更强的诱导胰腺癌细胞凋亡的能力。我们可以采用同样的策略,选用对肿瘤干细胞有靶向性的病毒载体, 同时携带双基因, 诱导肿瘤干细胞进入凋亡程序, 直接清除该细胞群体。
(5)增加对放疗和化疗的敏感性
放疗和化疗是当前肿瘤治疗的常规手段。但是在连续多次治疗后, 肿瘤容易产生抗药性, 致使疗效欠佳。另外, 放化疗后肿瘤的复发也是临床治疗中的常见问题。一般而言, 放疗主要影响的是分裂期细胞而对非分裂期细胞作用不明显。然而,肿瘤干细胞分裂缓慢, 多处于非分裂期, 因此对放疗的反应不敏感。放疗结束后, 肿瘤干细胞重新增殖直接导致肿瘤的复发。
Bao等人经研究 发现, 胶质瘤干细胞与非干细胞样胶质瘤细胞相比, 能更有效的启动DNA损伤检验点反应, 减少射线对其造成的伤害。在实验中,他们又通过抑制检验点激酶Chk1和Chk2的活性, 成功的削弱了胶质瘤干细胞对射线产生伤害的抵抗能力, 恢复了其对射线的敏感性。
另一方面, 肿瘤干细胞对化学药物的多药抗药性(multi drug resistance, MDR)与BCRP1、MGMT, 抗凋亡蛋白和MDR相关蛋白的高表达有密切关系。通过抑制这些抗性相关蛋白的表达,可以增强肿瘤干细胞对药物的敏感性, 提高其在化疗中的死亡率。可见,通过消除肿瘤干细胞对射线和药物的抗性这一策略, 仍然能够依靠放化疗这些传统的治疗手段来有效的清除肿瘤干细胞。
十、靶向肿瘤代谢的策略
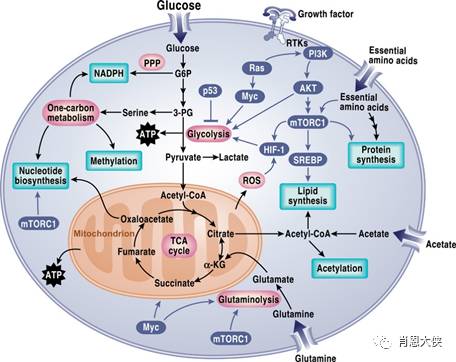
1、肿瘤无氧糖酵解途径的异常和调节,PKM2为关键分子
肿瘤细胞的能量供应主要依赖于无氧糖酵解,而丙酮酸激酶(PK)是这一过程中的限速酶,有两个异构体,分别为PK-M1和PK-M2,其中前者较后者有更高的活性。然而,既往研究显示,肿瘤细胞中活性较低的PK-M2呈高表达,而非活性较高的PK-M1。
美国学者孔(Kung)等通过采用PK-M2的异构激活剂AGX-257作用于多种肿瘤细胞,发现PK-M2的活性提高后肿瘤生长受到抑制;而通过对AGX-257作用前后进行对比发现,活性低的PK-M2无法将糖酵解途径中的中间体全部转化为丙酮酸,导致这些中间体合成丝氨酸进入氨基酸代谢途径,从而促进了肿瘤的生长。PK-M2激活剂可以作为肿瘤联合治疗的一部分。本研究不仅解释了肿瘤选择性高表达活性低PK的原因,更为重要的是,还为肿瘤的靶向治疗提供了新的治疗策略。
2、线粒体功能异常的调控作用。
线粒体是正常细胞的主要供能单位,而肿瘤细胞即使在有氧的条件下也以无氧糖酵解为主要的获能方式(即Warburg效应)。另外,肿瘤细胞中的线粒体数量并没有明显减少,提示肿瘤细胞的线粒体功能是异常的。国立卫生研究院(NIH)在内的一个联合研究团队,通过建立不同背景的线粒体和细胞核融合模型,发现功能异常的线粒体具有促进细胞生长和克隆形成的能力,而正常细胞的线粒体则不具备这一功能。
来自美国霍华德-休斯医学研究所的肖(Shaw)等发现,肿瘤细胞内的功能异常线粒体存在自嗜障碍,其主要的调节因子是ULK1(一种丝/苏氨酸蛋白激酶),ULK1上调可促进线粒体的自嗜,而ULK1的上游调节因子是LKB1。有趣的是,在非小细胞肺癌中,LKB1的突变率高达30%,且常与KRAS突变并存。研究者通过建立LKB1缺陷动物模型进行药物筛选,发现一种线粒体复合体Ⅰ的抑制剂phenformin对LKB1缺陷及突变型肿瘤细胞具有杀伤作用。此外,该研究团队还进一步构建了LKB1和KRAS双突变模型,发现phenformin同样有效,且可以逆转RAS-MEK抑制剂的耐药。清除肿瘤细胞的线粒体可能成为靶向治疗的新手段,其中靶向突变型的LKB1可能成为肺癌新的治疗手段。
3、中间体及其代谢物的调节作用
肿瘤细胞长期处于应激的微环境中,在代谢过程中会产生一系列活性氧簇(ROS),包括O2-、H2O2、HO2、OH等。ROS具有双向调控肿瘤细胞凋亡和增殖的作用,中、高浓度的ROS可以诱导细胞凋亡和死亡,而低浓度ROS能够影响一系列信号传导途径,促进细胞增殖和分化,但其中的机制尚不明确。美国芝加哥大学海(Hay)等发现,烟酰胺腺嘌呤二核苷酸磷酸(NADPH)是调整ROS浓度的关键点。提示检测并调整NADPH的浓度,有可能成为新的抗肿瘤治疗手段。
肿瘤代谢途径异常主要是由于两方面原因。第一,表达代谢途径中限速酶的基因突变或扩增,如胶质瘤和急性白血病中IDH1和IDH2基因出现突变,遗传性副神经节瘤中SDH基因功能缺失性突变,肾癌中FH基因功能缺失突变导致HIF-1α的稳定,以及黑色素瘤和乳腺癌中PHGDH的扩增和过表达。第二,控制代谢途径的癌基因和抑癌基因出现变异,例如PI3K/AKT/mTOR和AMPK的活化可以增加葡萄糖摄取、脂质合成及抑制自嗜,p53活化可诱导自嗜并增强线粒体的代谢功能,myc扩增或过表达可增加肿瘤细胞对谷氨酸的利用等。
因此,阻断代谢途径中的关键限速酶或者干预其上游的癌基因或抑癌基因均可在一定程度上抑制肿瘤的发展。目前在临床应用中,单一信号通路的靶向治疗效果常不理想,旁路途径的激活是重要原因之一。而克服旁路激活导致的耐药,目前多采用横向联合多条信号通路抑制剂的方式。由于各条信号通路最终会通过调控下游的代谢来影响细胞行为,因此纵向联合信号通路和代谢途径抑制剂亦可能产生增强的抗肿瘤效果。然而,代谢途径中存在不同的代谢通路,不同类型或不同状态的肿瘤细胞可能依赖于不同的代谢通路,因此发现肿瘤特异的代谢通路及其中的关键限速酶就成为肿瘤代谢学的重点和难点。
十一、针对端粒末端转移酶的靶向治疗
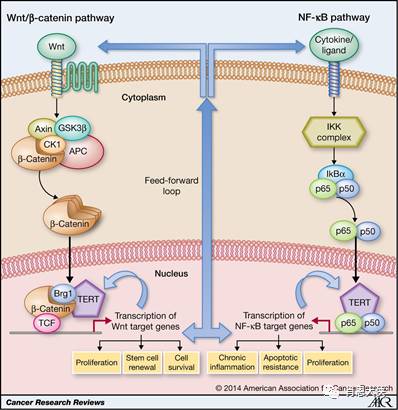
端粒末端转移酶和端粒在细胞衰老或永生化过程中起决定性的作用。端粒末端转移酶活性的再活化与肿瘤的生长有关,是恶性肿瘤共有的一个特征。因此,一些临床前及临床研究证据认为端粒末端转移酶有望成为肿瘤治疗的新靶点。最近,Marian 等用端粒末端转移酶抑制剂 GRN163L 靶向恶性胶质瘤(glioblastoma, GBM)干细胞,体外及体内实验均显示出良好的治疗效果。
GRN163L 能够使 GBM 干细胞端粒缩短、降低增殖率,导致干细胞死亡。GRN163L 与替莫唑胺及放射治疗联合应用能够发挥协同作用,更有效地抑制 GBM 干细胞的生长。发挥协同作用的机制:一方面,在端粒末端转移酶缺失的情况下,射线和替莫唑胺导致的 DNA 断裂不能被修复;另一方面,替莫唑胺能够启动恶性胶质瘤细胞的自溶作用,而 GRN163L 能够使该作用进一步加强。此外,GRN163L 能够通过血-脑脊液屏障,克服了常规抗肿瘤药物的不足。
十二、miRNAs与肿瘤靶向治疗
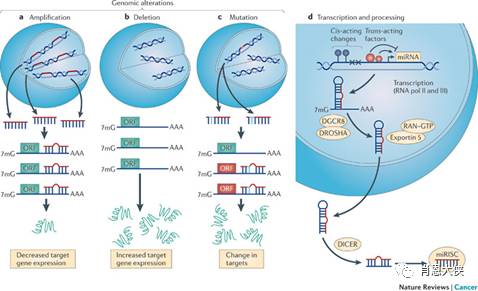
miRNAs是真核生物细胞中固有的一类长度约为22个核苷酸且不编码蛋白的小分子RNA。miR2NA在转录后水平负性调控靶基因的表达,通过与mRNA3’端非编码序列的结合或降解mRNA或抑制mRNA转录后的翻译。miRNAs广泛参与动植物生命活动的调控,如生长发育、营养物质的代谢和激素的分泌等。近年的研究表明,其不仅在肿瘤的发生和发展中发挥重要作用,在干细胞的自我更新和多向分化方面也起着重要作用。用生物信息学的方法发现,一些调控干细胞自我更新的因子,如Notch和PTEN,是miRNAs的靶基因,虽然这些结果尚待实验的证实,而且miRNAs调控干细胞自我更新的机制也需进一步深入的研究。
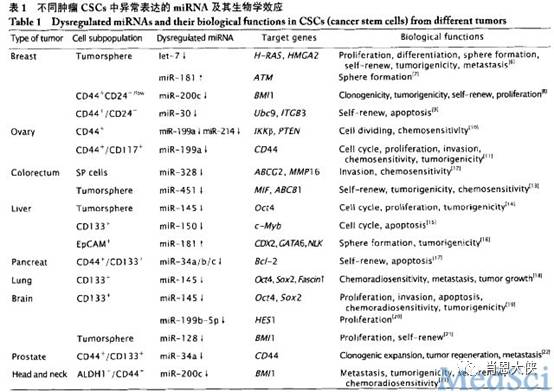
Hatfield等发现,在Dicer21(miRNAs生成过程中所必需的一种核糖核酸酶)突变的果蝇GSC细胞中,大部分细胞都停滞在细胞周期G1到S的转变期中,说明干细胞通过G1/S期转变需要miRNAs的作用,miRNAs在干细胞自身稳态的维持中发挥着重要的作用。仅在肿瘤干细胞中就有大量的miRNA表达,如下表;
十三、其它靶点
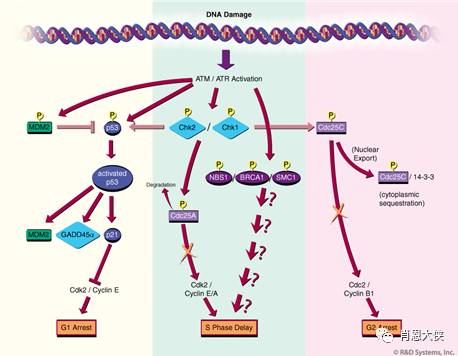
目前,针对上述多条通路中一些关键分子的抑制剂研究都有不同程度的进展。如O6-苄基鸟嘌呤( O6-BG)及其多个衍生物能特异抑制DR通路中O6-烷基鸟嘌啉-DNA 烷基转移酶(AGT),目前正在作为耐药逆转剂与甲基化剂合用进行临床试验。还有靶向抑制BER通路中的多聚(ADP-核糖)聚合酶(poly[ADP- ribose]polymerase,PARP)的AG014699、NU1025、AG14361和INO-1001分别处于Ⅰ~Ⅲ期临床实验。
这些抑制剂与烷化剂、拓扑异构酶I抑制剂、顺铂或γ-射线联合用于肿瘤细胞或者小鼠移植瘤,能分别增加后者引起的DNA损伤、细胞生长抑制和荷瘤小鼠的存活时 间。现今,关注最多的是修复DSBs的HR和NHEJ通路,分别在这两条通路中扮演关键角色的ATM和DNA-PK都是PI3K激酶家族成员,前文已经提到的wortmannin、LY294002和咖啡因等PI3K家族的广谱抑制剂都能通过抑制这两条修复通路的活性起到放化疗增敏的作用。
另外,还有 ATP的竞争抑制剂KU-55933能显著抑制ATM活性,且作用明显强于对PI3K家族其它成员的抑制效果,能增强ATM高表达细胞株对离子辐射和 VP16、阿霉素、喜树碱等造成DSBs药物的敏感性。DNA-PK的选择性抑制剂IC87361、Nu7026、Vanillin和SU117582也 有相似的作用。
十四、结语
多个分子靶向药物的成功上市为新一代抗肿瘤药物的研发提供了乐观的前景。但是,必需看到靶向信号通路抑制剂只有在该信号通路高度激活的肿瘤上才产生更好地疗效,因此合理的临床设计是体现其疗效的必要前提。只有在合理的研究策略、全面的临床前评价和严谨合理的临床试验的共同配合下才能开发出更多有希望的靶点特异性抗肿瘤新药。
参考文献:
1.Lafferty-Whyte K, Mormeneo D, del Fresno Marimon M. Trial watch: Opportunitiesand challenges of the 2016 target landscape[J]. Nature Reviews Drug Discovery,2016
2.Dienstmann R, Rodon J, Serra V, et al. Picking the point of inhibition: acomparative review of PI3K/AKT/mTOR pathway inhibitors[J]. Molecular cancertherapeutics, 2014
3.Minucci S, Pelicci P G. Histone deacetylase inhibitors and the promise ofepigenetic (and more) treatments for cancer[J]. Nature Reviews Cancer, 2006.
4.Corcoran N M, Clarkson M J, Stuchbery R, et al. Molecular pathways: TargetingDNA repair pathway defects enriched in metastasis[J]. Clinical Cancer Research,2016
5.Kasinski A L, Slack F J. MicroRNAs en route to the clinic: progress invalidating and targeting microRNAs for cancer therapy[J]. Nature reviewsCancer, 2011
6.Li Y, Tergaonkar V. Noncanonical functions of telomerase: implications intelomerase-targeted cancer therapies[J]. Cancer research, 2014
肖恩大侠 华盖医疗基金董事总经理,北京。
毕业于北京大学工学院/医学院(医学硕士)、北京大学法学院(法学硕士)。资浅PE从业人士。躬耕于太湖,游学至未名,当过医生,学过法律,改行至风险投资,喜好读书写作,经史文集,现代科技都甚喜欢,偶尔有点愤青,时常有点较真,总是怀着理想,万一,实现了呢?
欢迎交流医疗、文化、消费,欢迎交流一级市场、二级市场,欢迎交流国内及海外并购(有标的需要一起合作)。
邮箱:[email protected]


欢迎联系我们![email protected]










