本研究通过培养组学方法,从健康人粪便样本中分离出超过6000株细菌,构建了包含1520个高质量单菌基因组草图的可培养基因组参考(CGR)数据集。这些基因组覆盖了人肠道的主要细菌门和属,包括至少264个新参考基因组,提高了宏基因组学分析的分辨率和功能注释的准确性。对38种重要菌的泛基因组分析揭示了不同菌门的泛基因组开合趋势和在各自核心和非核心基因组上功能富集的差异。这些成果将有助于人肠道宏基因组学分析和相关应用转化研究。
肠道微生物群在人体健康和疾病中有重要作用,但现有参考基因组代表性不足,限制了宏基因组学分析的高分辨率。
从健康人粪便样本中分离超过6000株细菌,构建包含1520个单菌基因组草图的CGR数据集,并进行功能注释和泛基因组分析。
CGR数据集覆盖了人肠道的主要细菌门和属,包括至少264个新参考基因组,提高了宏基因组学分析的分辨率和功能注释的准确性。
CGR数据集将有助于人肠道宏基因组学分析和相关应用转化研究,推动肠道微生物组研究的深入。

人类肠道培养细菌培养组的参考基因组集
人类肠道培养细菌的1520个基因组扩展微生物组功能分析
1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses
Nature Biotechnology
[IF:31.864]
2019-04-04 Articles
DOI: https://doi.org/10.1038/s41587-018-0008-8
全文可开放获取 https://www.nature.com/articles/s41587-018-0008-8.pdf
第一作者:Yuanqiang Zou, Wenbin Xue, Guangwen Luo, Ziqing Deng, Panpan Qin
1,2
通讯作者:Junhua Li(李俊桦 [email protected]), Huijue Jia(贾慧珏 [email protected]) & Liang Xiao(肖亮 [email protected])
1,2*
其它作者:Ruijin Guo, Haipeng Sun, Yan Xia, Suisha Liang, Ying Dai, Daiwei Wan, Rongrong Jiang, Lili Su, Qiang Feng, Zhuye Jie, Tongkun Guo, Zhongkui Xia, Chuan Liu, Jinghong Yu, Yuxiang Lin, Shanmei Tang, Guicheng Huo, Xun Xu, Yong Hou, Xin Liu, Jian Wang, Huanming Yang, Karsten Kristiansen
作者单位:
1
华大基因(BGI-Shenzhen, Shenzhen, China)
2
国家基因库(China National Genebank, BGI-Shenzhen, Shenzhen, China)
3
丹麦哥本哈根大学(Laboratory of Genomics and Molecular Biomedicine, Department of Biology, University of Copenhagen, Copenhagen, Denmark)
热心肠日报
https://www.mr-gut.cn/papers/read/1044065063
华大基因团队:1520个人肠道单细菌参考基因组,助力菌群研究
创作:mildbreeze 审核:mildbreeze
2019年03月01日
-
用培养组学分离>6000个人粪便菌株,得到1520个高质量单菌基因组草图(338个种),构成可培养基因组参考(CGR)数据集;
-
包括人肠道的主要细菌门和属,含至少264个新参考基因组,有不少低丰度菌,丰富了现有参考基因组,提高了宏基因组学分析的分辨率(读段比对率和SNP分析等);
-
对CGR的功能注释加深了对肠道菌功能的认知;
-
对38种重要菌的泛基因组分析,揭示不同菌门的泛基因组开合趋势和在各自核心和非核心基因组上功能富集的差异。
主编推荐语: 华大基因的肖亮、贾慧珏和李俊桦与团队的重要研究在Nature Biotechnology发表。该研究通过培养组学方法,从健康人粪便样本中获得1520个单菌基因组草图,对现有的人肠道细菌参考基因组有很大补充;并通过功能注释和泛基因组分析,加深了对肠道细菌功能和特点的了解。这些成果将助力人肠道宏基因组学分析和相关应用转化研究。
摘要
参考基因组对于人类肠道微生物组的宏基因组分析和功能表征至关重要
。我们展示了可培养基因组参考 (Culturable Genome Reference,CGR),它包含 1,520 个非冗余、高质量的基因组草图,这些基因组草图由从健康人类粪便样本中培养的 6,000 多种细菌生成。在选择涵盖人类肠道中所有主要细菌门和属的 1,520 个基因组中,
264 个未在现有参考基因组集中显示
。我们表明,参考细菌基因组数量的增加将宏基因组测序读数的比对率从 50% 提高到 >70%,从而能够更高分辨率地描述人类肠道微生物组。我们使用 CGR 基因组来
注释 338 个细菌物种的功能,显示了该资源在功能研究中的实用性
。我们还对
38 个重要的人类肠道物种进行了泛基因组分析,揭示了其核心基因组和可有可无的基因组之间功能富集的多样性和特异性
。
背景
人体肠道菌群是指居住在人体胃肠道中的所有微生物。肠道微生物群在人类健康和疾病中的多种作用已得到认可。宏基因组研究改变了我们对人类微生物群分类和功能多样性的理解,但
来自典型人类粪便宏基因组的超过一半的测序读数无法映射到现有的细菌参考基因组。缺乏高质量的参考基因组已成为对人类肠道微生物组进行高分辨率分析的障碍
。
尽管之前报道的整合基因集 (IGC) 已经启用了宏基因组、宏转录组和宏蛋白质组分析,但
组成分析和功能分析之间的差距只能由单个细菌基因组来填补
。样本间共变的基因可以聚类为宏基因组连锁群、宏基因组簇和宏基因组物种,其
注释取决于与有限数量的现有参考基因组的比对。其他基于宏基因组学的肠道微生物组分析——例如,单核苷酸多态性 (SNP)、插入缺失和拷贝数变异——依赖于参考基因组的覆盖率和质量
。
尽管测序的细菌和古细菌基因组的数量迅速增加,但肠道细菌的参考基因组代表性不足。据估计,美国国家生物技术信息中心 (NCBI) 数据库中 <4% 的细菌基因组属于人类肠道微生物群。相反,重点一直放在临床相关的病原菌上,这些细菌在微生物数据库中过多。人类微生物组计划 (HMP) 于 2010 年报告了人类微生物群的 178 个参考细菌基因组的第一个目录。迄今为止,HMP 已对从人体部位培养的 2,000 个微生物基因组进行了测序,其中 437 个是肠道微生物群。2017 年 9 月 8 日访问的数据)。然而,参考肠道细菌基因组的数量还远未饱和。
我们提供了人类肠道细菌(命名为 CGR)基因组的参考目录,该目录是通过从健康个体的粪便样本中分离出超过 6,000 株细菌而建立的。CGR 包含 1,520 个非冗余、高质量的细菌基因组草图,为肠道微生物组贡献了至少 264 个新的参考基因组。纳入 CGR 基因组后,选定的宏基因组数据集的
比对率从大约 50% 提高到 70% 以上
。除了改进宏基因组分析外,CGR 还
将以高分辨率改进肠道微生物群的功能表征和泛基因组分析
。
结果
扩展人类肠道细菌基因组集
Expanded catalog of gut bacterial genomes
从155个人粪便使用11种培养基厌氧条件下,获得6487个细菌培养物(附图1)。值得注意的是,超过一半的分离株是从 MPYG 培养基中培养出来的。16S扩增子测序获得1759个非冗余。全基因组测序。发现
104个包括大于1个基因组
。多个基因组在GC含量和测序深度角度进行拆分。这些组装分析为212个基因组(分箱)。共获得1867个基因组,1520(81.4%)达到HMP的高质量标准,即95%完整性,<5%污染率使用CheckM评估。基因组大小范围0.2~7.9,GC含量范围26.56%~64.28%。
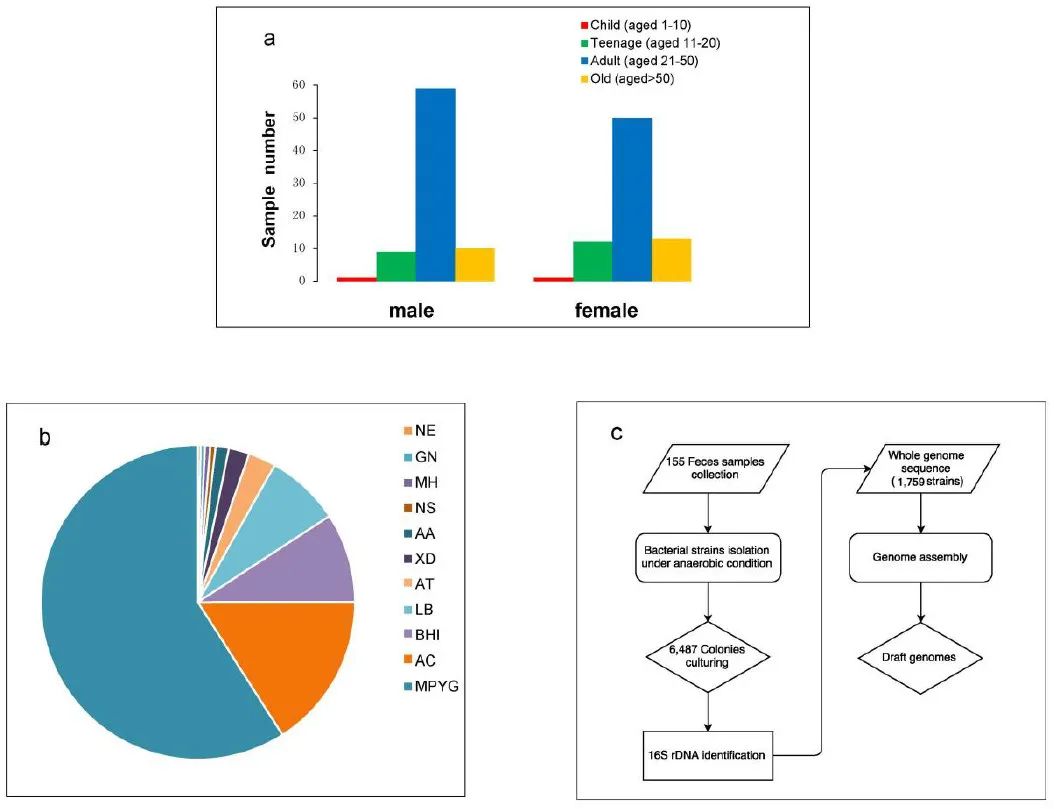
附图1. 肠道菌群的培养和基因组测序
(a) 155个我粪便样本按年龄和性别分类;(b) 厌氧条件下不同培养基因的分离数量;(c) 培养测序的流程
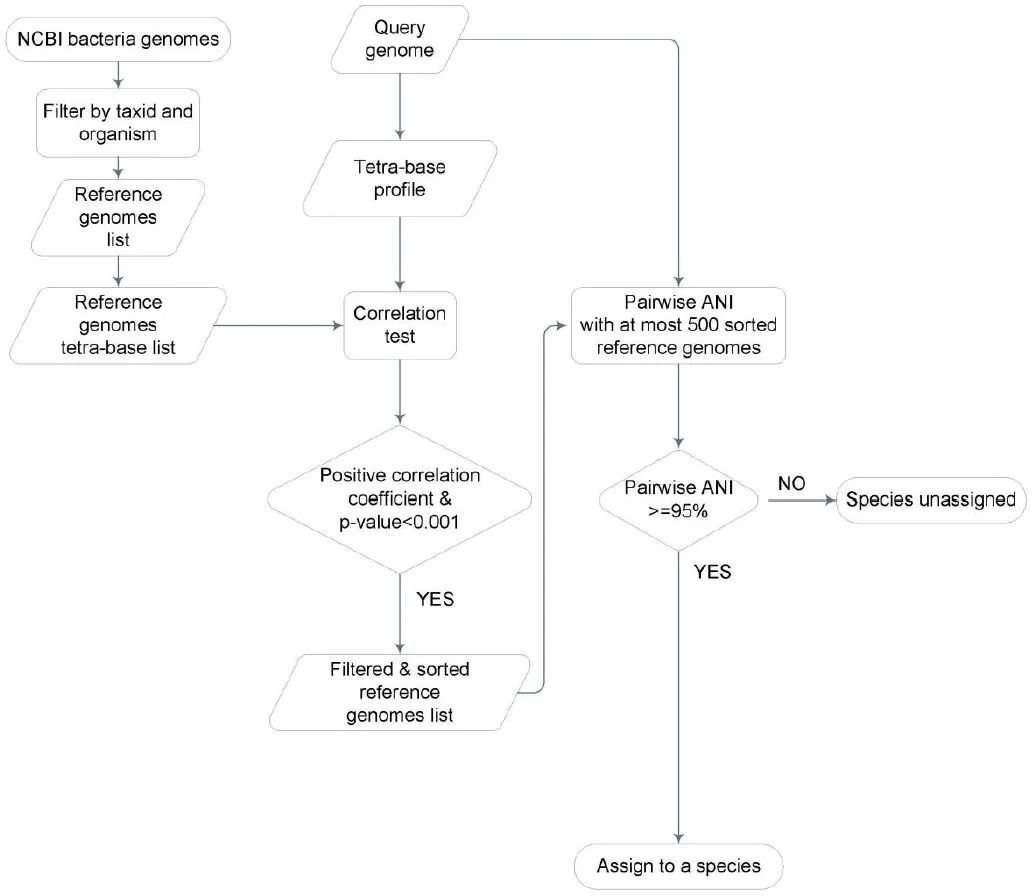
附图2. 测序基因组的物种注释
基于ANI的流程,采用POCP补充分析(目前用GTDB更方便)
CGR 的分类注释是使用自建的、高效的基于 ANI 的流程进行(补充图 2)。1,520 个高质量基因组被分为 338 个物种水平的簇(
ANI ≥ 95%,对应于 70% DNA-DNA
杂交的物种描述),涵盖了人类肠道微生物群的所有主要门,包括厚壁菌门(211 个簇) ,796 个基因组),拟杆菌(60 个簇,447 个基因组),放线菌(54 个簇,235 个基因组),变形杆菌(10 个簇,36 个基因组)和梭杆菌(3 个簇,6 个基因组)(图 1a 和补充表 5)。在这 338 个簇中,
134 个簇(对应于 264 个基因组)没有被 NCBI 中的任何现有参考基因组注释(图 1a),并且 50 个簇不属于任何已测序的属
(补充表 5)。为了证实 CGR 中新物种的存在,我们使用 16S rRNA 基因分析进行了额外的分类鉴定。如果一个物种的
16S rRNA 基因序列与 EzBioCloud 数据库中的已知物种具有 < 98.7% 的相似性,则该物种被认为是新物种
(参见方法)。总体而言,我们确定了 350 个不同的细菌物种(基于操作分类单位),包括 149 个候选新物种,其中 42 个代表候选新属。这些结果强调了 CGR 提供的个体参考基因组的价值。
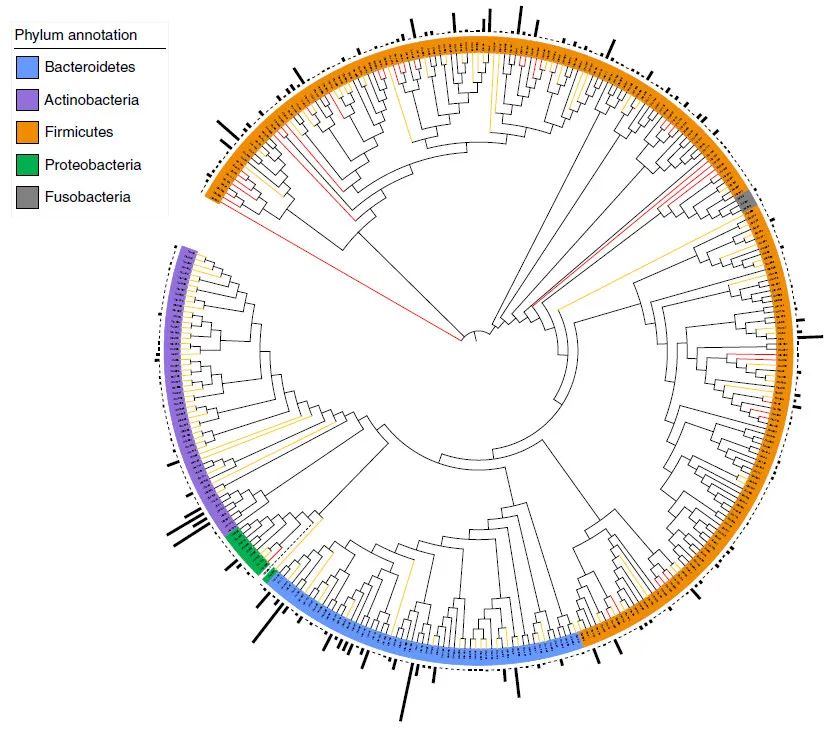
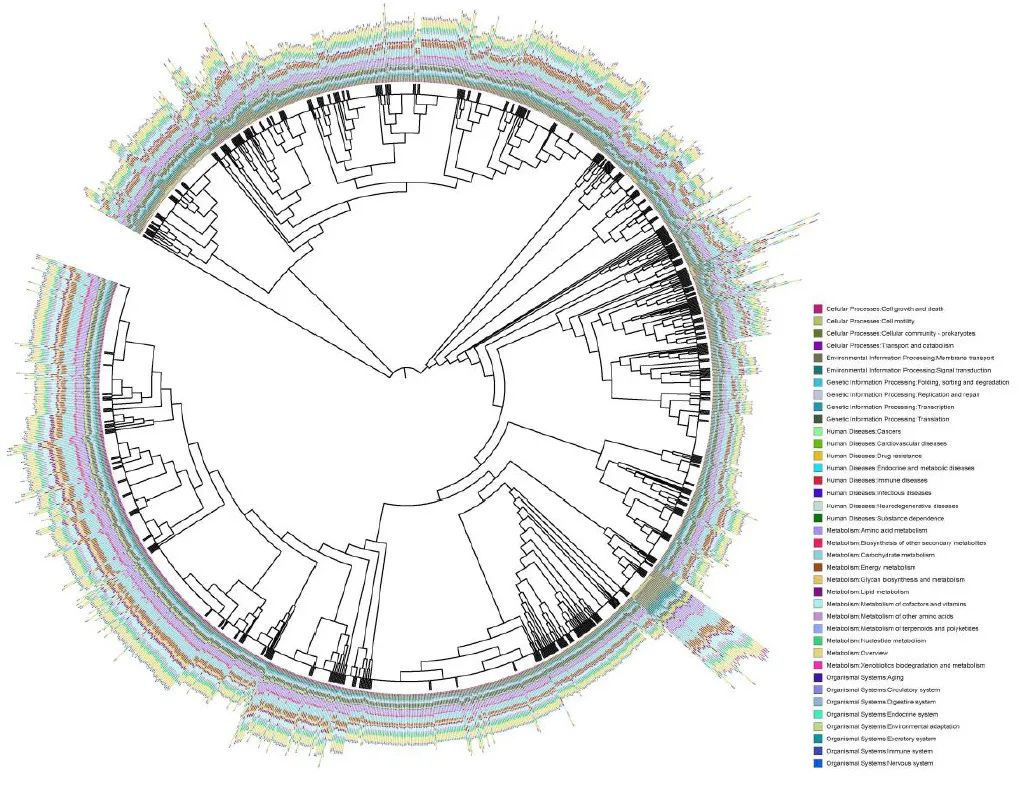
图 1:基于全基因组序列的 1,520 种分离肠道细菌的系统发育树。
Fig. 1: Phylogenetic tree of 1,520 isolated gut bacteria based on whole-genome sequences.
CGR 中的 1,520 个高质量基因组根据它们的全基因组序列分为 338 个物种级簇(ANI ≥ 95%)。厚壁菌门的细菌种类为橙色;拟杆菌,蓝色;变形菌,绿色;放线菌,紫色;梭杆菌,灰色。新属和新种分别用红色和橙色分支突出显示。
最外层的条形表示每个集群中存档的基因组数量
。硒化根瘤菌 ATCC BAA 1503 被用作系统发育分析的外群。
尽管在属水平上个体微生物群存在差异,但 CGR 确定了具有广泛多样性的细菌种群,涵盖了中国肠道微生物群中九个核心属中的八个。与来自人类胃肠道的 1000 个培养细菌物种的先前测序物种相比,80 多个物种是新的(补充图 3a)。此外,根据 IGC,CGR
成功鉴定了 38 个低相对丰度( < 1%)的属
,这是一个大型参考基因目录,来自来自三大洲个体的约 1,250 个宏基因组样本(补充图 1)。3b)。其中,共鉴定出7个属、20多个基因组(双歧杆菌、柯林斯菌属、粪杆菌属、多雷亚菌属、链球菌属、普氏菌属和副杆菌属)。CGR 还鉴定了 IGC未检测到的另外 9 个属(Butyricicoccus、Butyricimonas、Catenibacterium、Dielma、Erysipelatoclostridium、Megamonas、Melissococcus、Peptoclostridium 和 Vagococcus)(补充图 3b)。这些结果强调了 CGR 对现有肠道细菌全基因组数据库的贡献。
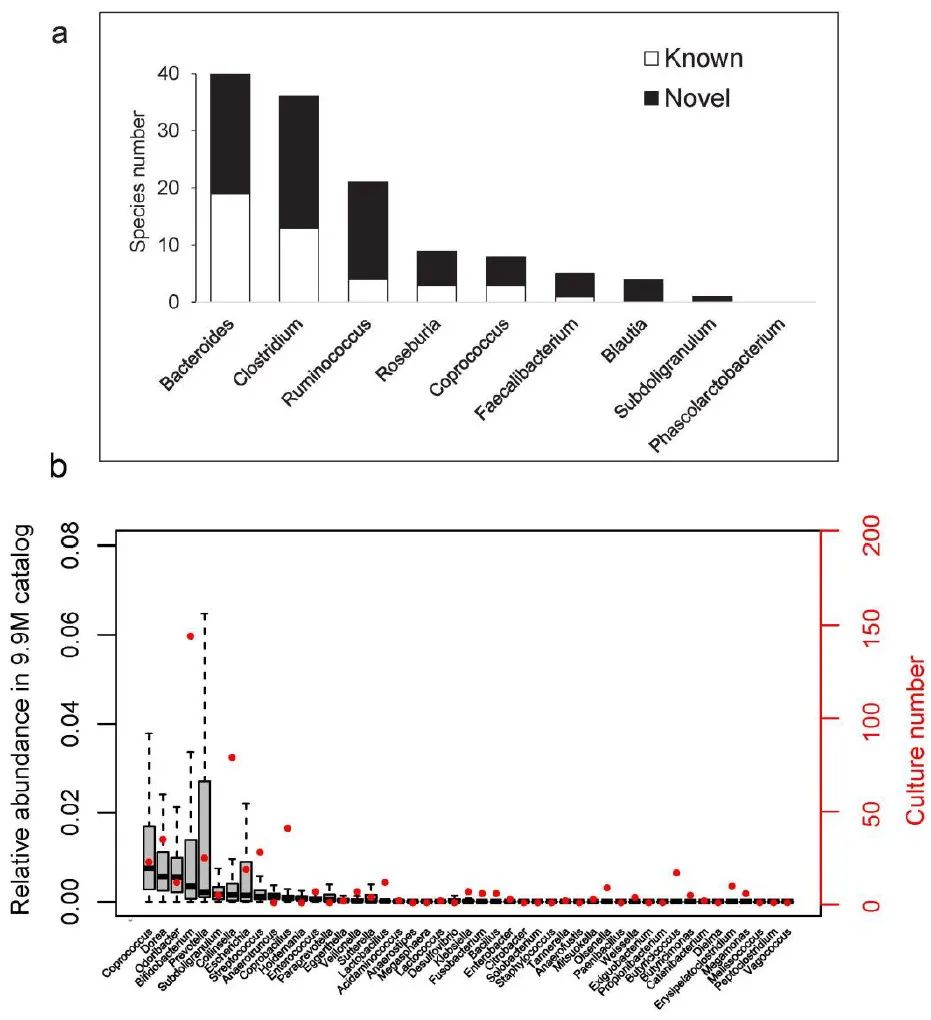
附图3. CGR 中肠道细菌基因组的多样性和新颖性
(a) 己知和新发现的种比例:CGR中的属于中国人肠道微生物群9个核心属的细菌种类数。将CGR的细菌种类与先前报道的人类胃肠道中的 1,000 种培养细菌种类进行比较,已知种类以白色显示,新种类以黑色显示。
( b )相对丰度和培养的数量。在 CGR 中鉴定的低丰度(<1%)肠道细菌属。根据之前的 IGC 研究,灰色框表示来自 1267 个样本的
每个属的相对丰度
。
红点表示本研究存档的每个属的物种数
。每个箱线图说明了估计的中位数(中心线)、上下四分位数(箱限)、1.5 × 四分位距(须线)。
改进宏基因组和 SNP 分析
Improvement in metagenomic and SNP analyses
用于宏基因组序列比对的现有参考基因组远未饱和。例如,最近一项研究中使用的细菌和古细菌的基因组或基因组草图允许绘制粪便宏基因组中不到一半的序列。为了说明 CGR 对宏基因组分析的价值,我们使用先前有或没有 CGR 的宏基因组数据进行了序列比对。对于中国样本,使用 IGCR 数据集(来自 IGC 的 3,449 个参考基因组)的原始研究中的
读长比对为 52.00%,在包含 CGR 数据集后显着提高至 76.88%
(图 2a 和补充表 6) )。由于 CGR 中的所有样本都来自中国,因此可以合理地假设该基因组数据集对中国粪便宏基因组有重大贡献。为了评估 CGR 对非中国宏基因组图谱的贡献,我们使用来自美国、西班牙和丹麦粪便样本的宏基因组数据进行了类似的分析。值得注意的是,
这些样本的宏基因组读长比对比率均大幅增加(图 2a),尽管与中国样本相比程度较小
(补充图 4a)。中国和非中国样本比对率的提高表明 CGR 涵盖了这些国家之间人们共享的相当数量的肠道细菌物种。为了揭示 CGR 实现的基因和蛋白质多样性的改善,我们比较了基于先前 IGC 研究中使用的基因组和添加 CGR 后的基因和蛋白质累积曲线(补充图 4b,c)。基因和蛋白质家族的数量随着前 1,500 个基因组的加入而增加,但或多或少地稳定在大约 3,000 个基因组。我们的 CGR 基因组的添加导致添加的基因和蛋白质家族的数量随着基因组数量的变化而大幅增加。通过包含 CGR,
总共添加了 373,555 个基因簇和 149,945 个蛋白质簇,对应于已知基因和蛋白质序列多样性分别增加了 22% 和 16%
。
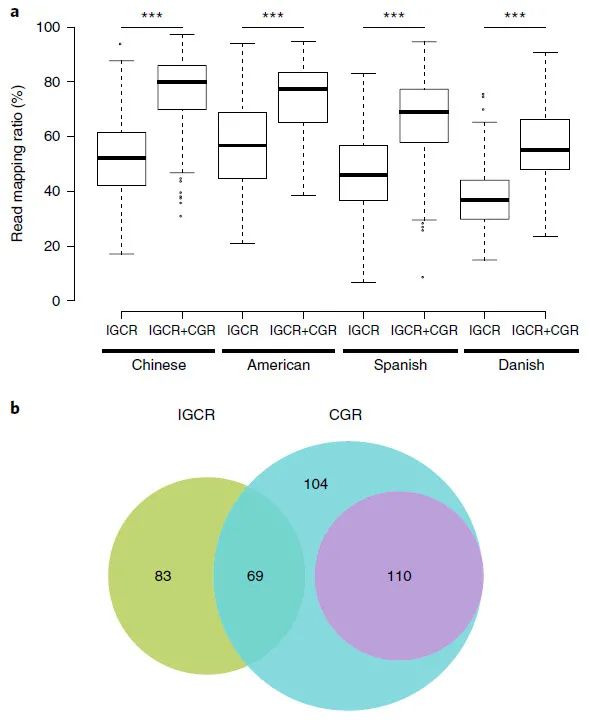
图 2:CGR 对宏基因组和 SNP 分析的贡献。
Fig. 2: Contribution of CGR to metagenomic and SNP analyses.
a,在来自中国(n = 368,P = 6 × 10-78)、美国(n = 139,P)的粪便样本中,CGR(IGCR + CGR)显着提高了先前宏基因组分析(IGCR)的读数映射率 = 2 × 10−17)、西班牙(n = 320,P = 4 × 10−50)和丹麦(n = 109,P = 4 × 10−17)个体。改善的显着性由两侧 Wilcoxon 秩和检验确定。IGCR,IGC 研究中使用的 3,449 个参考基因组6;CGR,本研究中生成的 1,520 个参考基因组。每个箱线图说明了读取映射比率的估计中位数(中心线)、上下四分位数(箱限)、1.5 × 四分位距(须线)和异常值(点)。b,先前研究(IGCR,绿色)和当前研究(CGR,蓝色)中生成的用于 SNP 分析的参考基因组。CGR 中未分类的参考基因组物种以紫色突出显示。
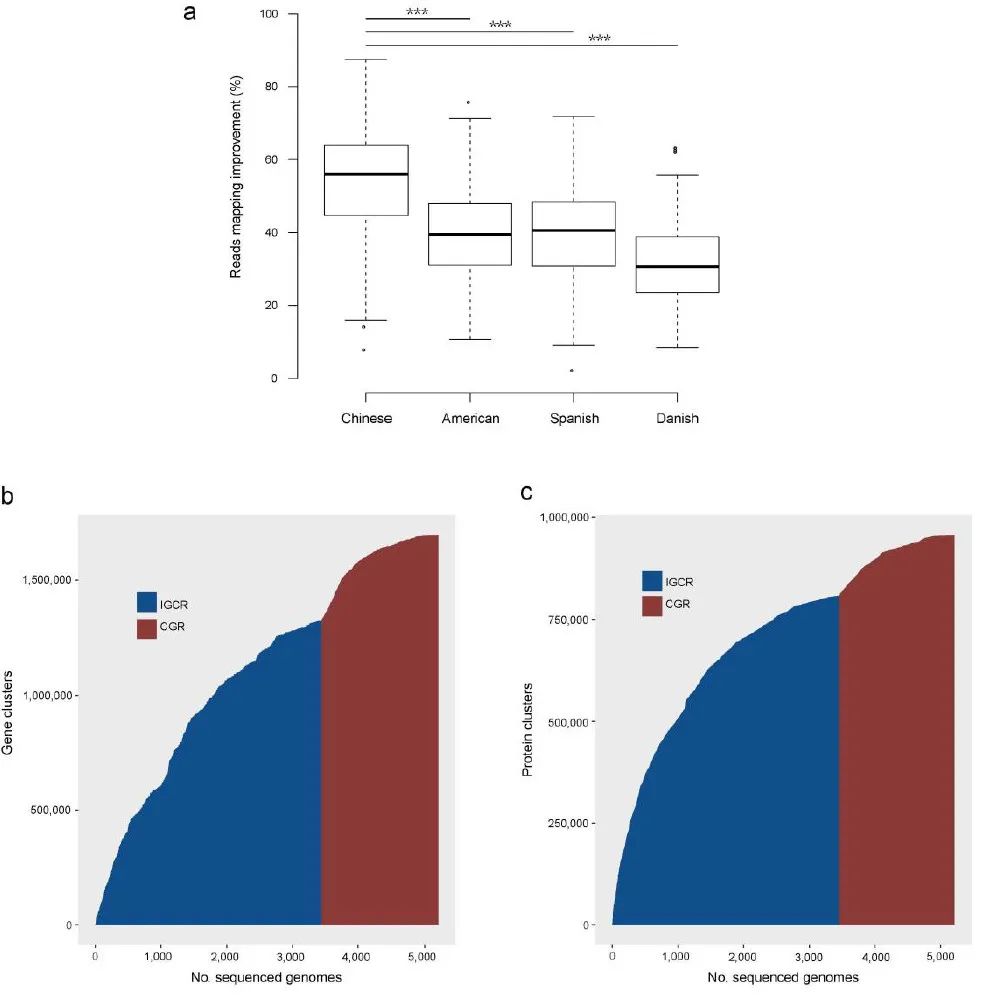
附图4. CGR 宏基因组分析的改进
(a) CGR 宏基因组分析中读长比对率的提高(与图 2a 相关)。改善百分比由以下公式计算:(CGR-ICG)/(100-ICG)。中国 (n=368) 的改善百分比显着高于美国 (n=139, P=8×10
-20
)、西班牙语 (n=320, P=9×10
-33
) 和丹麦语 (n= 109, P=2×10
-31
) 个个体。改善的显着性由未配对的 Wilcoxon 秩和检验(双边)确定。ICG 代表从 3,449 个参考基因组(图 2a 中的 ICGR)计算的读长比对率,CGR 代表通过添加 1,520 个参考基因组(图 2a 中的 ICGR+CGR)计算的读长比对率。每个箱线图说明读长比对率的估计中位数(中心线)、上下四分位数(箱限)、1.5 × 四分位距(须线)和异常值(点)。(b)(c) 基因和蛋白质序列多样性CGR 增加。从 ICGR(蓝色)和 CGR(红色)添加的基因组中新基因家族(b)和蛋白质家族(c)的数量增加。
为了进一步说明 CGR 的效用,我们使用它来分析来自 TwinsUK 注册中心的 250 个样本队列中的肠道微生物组 SNP。我们从 CGR 生成了一组新的 282 个非冗余代表性基因组(参见方法、补充图 5 和补充表 7),其数量几乎是原始 TwinsUK 分析中使用的 152 个参考基因组的两倍。为了突出通过分析现有基因组和 CGR 基因组确定的新参考基因组,我们对 282 个基因组与先前报告的 152 个基因组进行了基于 ANI 的比对。在新增的 192 个参考基因组中,85 个为分类物种,107 个为未分类物种(图 2b)。在瘤胃球菌属中发现了高 SNP 密度。CAG:108 (Clu 21)、未分类的厚壁菌门 (Clu 157)、直肠真杆菌 (Clu 6)、大肠杆菌(Clu 22) 和瘤胃球菌属。CAG:57 (Clu 19),表明这些物种的基因组存在高度变异,而加氏乳杆菌 (Clu 241)、粪肠球菌 (Clu 316)、杜兰肠球菌 (Clu 274) 和变形链球菌 (Clu 217) 显示出较低的 SNP 密度。共鉴定了 914 万个 SNP。由于 CGR 中
新添加的高质量参考基因组,某些物种的 SNP 数量有所增加
。我们得出结论,CGR 是宏基因组研究的宝贵资源,因为它可以显着提高宏基因组分辨率。
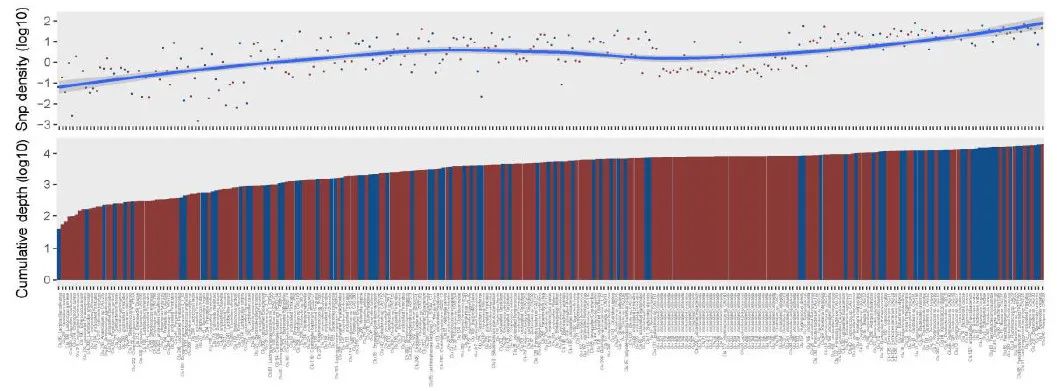
附图5. 282 个参考基因组中的 SNP 密度,在来自 TwinsUK 注册中心的 250 个样本中累积覆盖率至少为 10 倍。
参考基因组根据累积覆盖率排序,本研究生成的新参考基因组以红色突出显示。
肠道微生物组细菌的功能
Functions of gut microbiome bacteria
为了更好地阐明肠道微生物群的功能,我们使用 KEGG(京都基因和基因组百科全书) 注释了 1,520 个 CGR 基因组中的基因功能。
KEGG 2 级的功能通路表明,在所有分离的菌株中,参与碳水化合物和氨基酸代谢的通路都很丰富
,这表明这些是肠道微生物群的核心功能(补充图 6)。我们还分析了
KEGG 3 级通路,并专注于那些在门或属水平上富集的通路
(图 3a)。我们发现脂多糖生物合成 (ko00540) 基因广泛分布于革兰氏阴性菌的主要门:梭杆菌门、拟杆菌门和变形杆菌门中。参与聚糖降解的基因(ko00531 和 ko00511)在拟杆菌门的基因组中很丰富。这一观察结果与拟杆菌属成员是重要的人类肠道共生体的观点一致,它们有助于降解饮食和肠道粘膜中的聚糖。拟杆菌的成员还拥有参与鞘脂代谢 (ko00600)、鞘糖脂生物合成 (ko00601、ko00603 和 ko00604) 和类固醇激素生物合成 (ko00140) 的高比例基因。鞘脂和激素生物合成在真核细胞中无处不在,但在大多数细菌中不存在。这些结果表明拟杆菌的成员不仅参与肠道中的能量代谢,而且还可能在哺乳动物细胞中的鞘脂和激素信号传导中起作用。
变形菌门在参与异生物质降解的基因 (ko01220) 中表现出相对较高的丰度,这可能有助于肠道中环境化学物质和药物的降解
。
附图6. CGR 中 1,520 个基因组的功能注释
基因组中的基因功能使用KEGG通路进行注释,图中显示了2级功能。最外层的堆栈条代表每个基因组中具有给定功能的基因数量。系统发育树根据图 1 绘制。
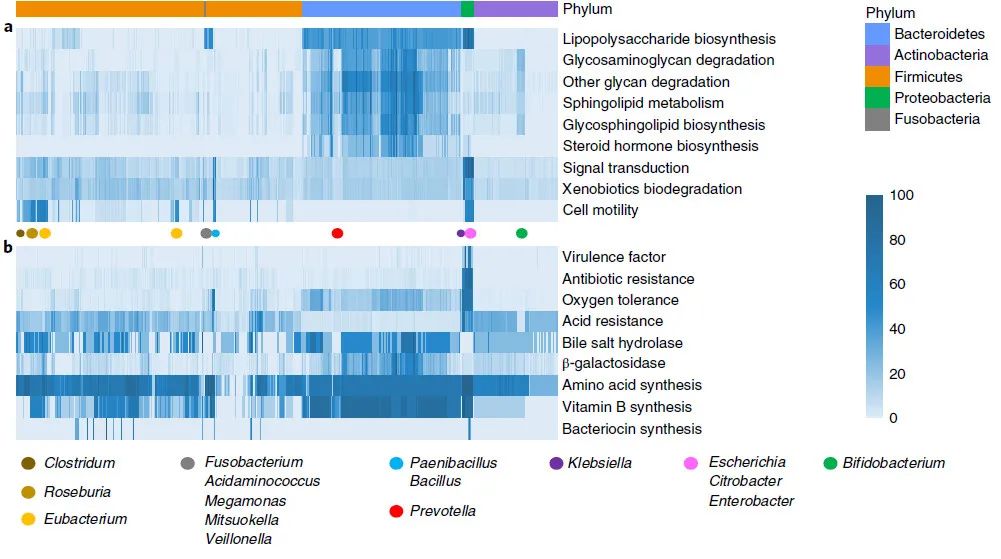
图 3:肠道微生物群的功能概述
Fig. 3: Functional landscape of gut microbiota.
CGR 的 1,520 个基因组中所列功能的基因丰度由热图中的颜色深度表示。所列功能在特定门或属 (a) 中丰富,或者可能对人类健康产生有害或有益影响 (b)。细菌种类根据图1中的系统发育树进行排序。系统发育树中门和属的相对位置分别用彩色带和点表示。
信号转导系统
(双组分系统,ko02020)和
异生物质降解
(KEGG 2 级通路)在类芽孢杆菌属、芽孢杆菌属、克雷伯菌属、埃希氏菌属、柠檬酸杆菌属和肠杆菌属中普遍存在,它们也存在于环境生态位,如土壤和 水。丰富的信号转导和异生物质降解系统使这些属能够感知和响应自然环境中存在的各种压力和有毒物质。
细胞运动性
(趋化性,ko02030;鞭毛组装,ko02040)在罗斯氏菌属、类芽孢杆菌属、芽孢杆菌属、埃希氏菌属、柠檬酸杆菌属和肠杆菌属中是保守的,但在梭菌属和真杆菌属内有所不同。
接下来,我们研究了 KEGG 数据库中注释的功能和通路,但未归类为 KEGG 通路(图 3b 和补充表 9)。
毒力因子和抗生素抗性基因分别使用毒力因子数据库 (VFDB) 和综合抗生素抗性数据库 (CARD)
进行注释。毒力因子和抗生素耐药性具有临床相关性,并且在变形菌门中含量丰富,这表明该门可能是机会性病原体的宿主。我们检查了与肠道细菌经常遇到的压力反应相关的基因分布:耐氧性和耐酸性。氧耐受性由编码过氧化氢酶和超氧化物歧化酶的基因数量反映,这两种解毒酶清除有氧呼吸过程中产生的活性氧。正如预期的那样,类芽孢杆菌属、芽孢杆菌属、克雷伯氏菌属、埃希氏菌属、柠檬酸杆菌属和肠杆菌属中的兼性厌氧菌更耐氧。除了先前报道的脆弱拟杆菌外,拟杆菌的其他成员也表现出中等的氧耐受性。值得注意的是,拟杆菌门和双歧杆菌属中的细菌通常缺乏耐酸基因,这表明基于这些生物的潜在益生菌口服后可能会在酸性胃环境中生存受损。最后,我们检查了 CGR 中可能对人类健康产生有益影响的六种细菌功能的分布。
氨基酸和维生素 B 合成基因
广泛存在于各种肠道细菌中,这表明肠道微生物群可能是素食中稀少营养素的替代来源。编码细菌胆汁盐水解酶的基因,在人类肠道中将初级胆汁酸转化为次级胆汁酸,在大多数肠道细菌中也普遍存在。编码 β-半乳糖苷酶的基因可能会减轻与乳糖不耐症相关的问题,在拟杆菌门中相对丰富。肠道细菌中参与
细菌素合成
的基因相对罕见,并且没有显示出门或属特异性分布。
肠道细菌的核心和泛基因组
Core and pan-genomes of underrepresented gut bacteria
我们对包含
10 个以上基因组的 36 个物种或簇
以及健康对照中富含的另外两个物种进行了泛基因组分析,与之前研究中的 2 型糖尿病患者相比,7,23,24,Fecalibacterium prausnitzii(簇 63 , 7 个基因组) 和产丁酸盐细菌 SS3_4 (簇 45, 9 个基因组)。这些簇涵盖了厚壁菌门、拟杆菌门、放线菌门和变形菌门(补充图 7a 和补充表 8a)。一个簇的
泛基因组可以定义为该簇内所有成员的核心基因和可有可无的基因(包括独特基因和辅助基因)的总和
。我们的泛基因组分析表明,
直肠真杆菌(第 37 簇)包含的核心基因比例最低(12%);其余基因属于附属基因组和独特基因组(分别为 38% 和 40%)
。相比之下,真杆菌 3_1(簇 6)包含最大比例的核心基因(53%)(补充图 7b)。泛基因组拟合曲线显示,拟杆菌中的大多数簇显示出“开放”泛基因组并且具有相对较大的泛基因组大小,其中普通拟杆菌在 14,970 个基因处具有最大的泛基因组大小(补充图 8 和 9和补充表 8b)。相比之下,放线菌门中的成员往往代表一个相对“封闭”的泛基因组,仅通过添加 CGR 基因组而略微扩大。这些结果表明肠道细菌基因组在拟杆菌门中是可变的,在厚壁菌门和变形菌门中的可变性较小,而在放线菌中则相当保守。
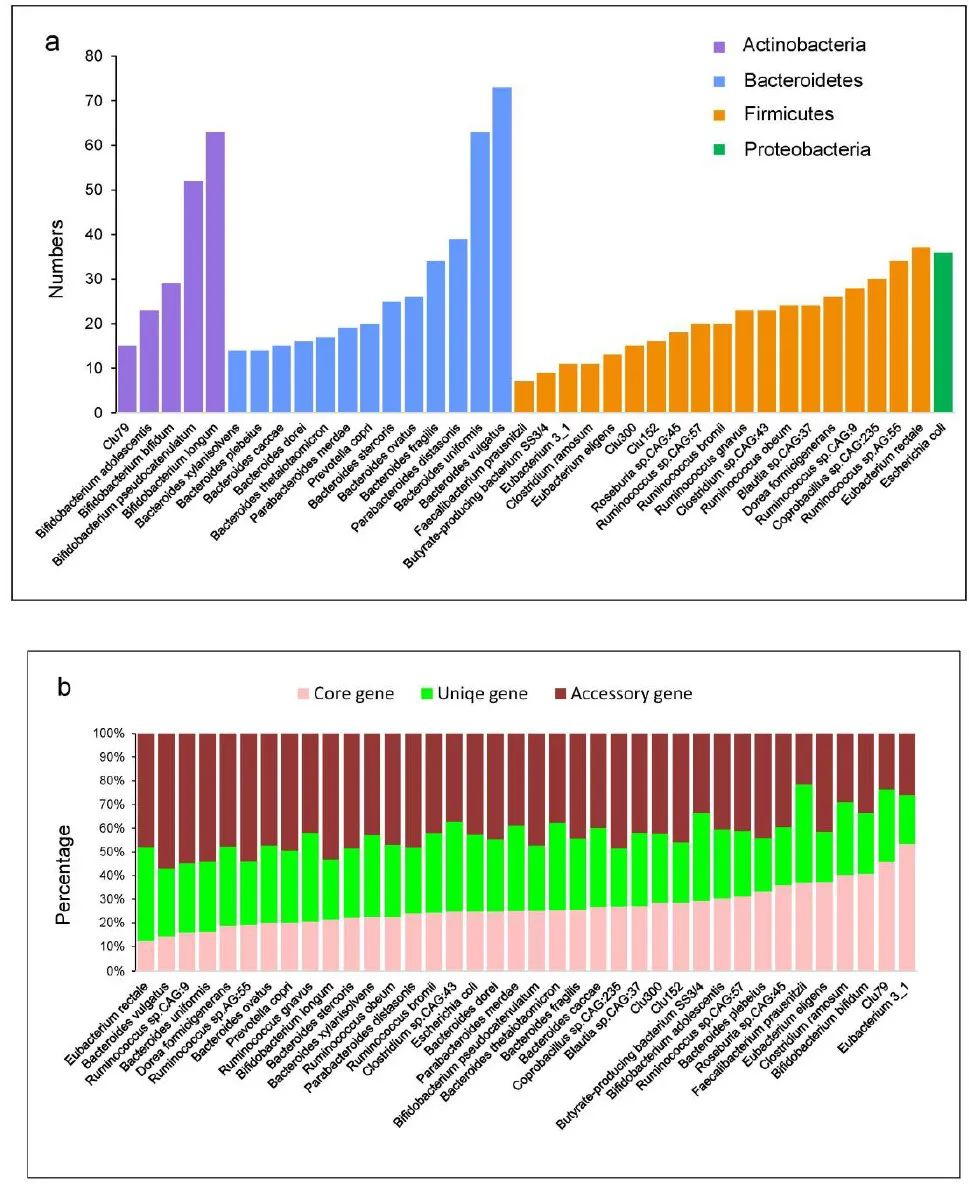
附图7. 38 个簇的泛基因组分析的统计数据。
(a) 泛基因组分析中使用的每个簇的基因组数量(按名着色,数量排序)。
(b) 38个簇基因组中核心基因、独特基因和辅助基因的组成。簇按核心基因的比例排序。
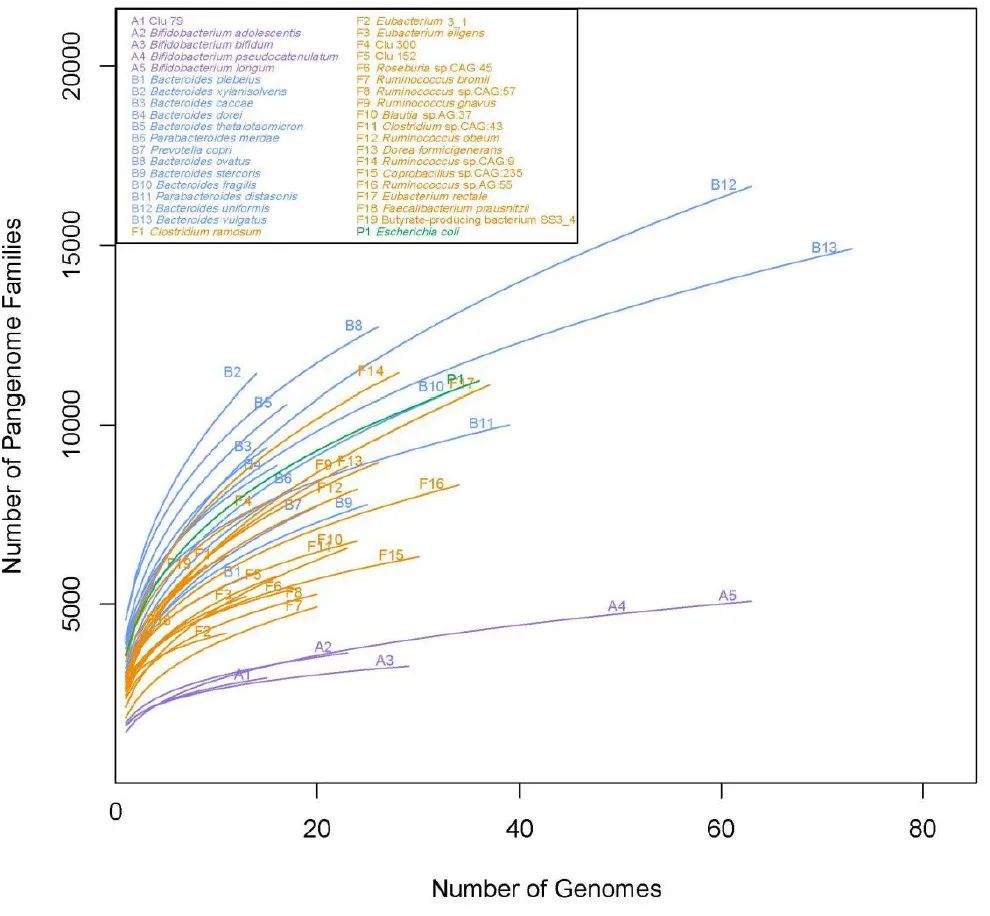
附图8. 38个簇的泛基因组拟合曲线
来自厚壁菌门(橙色)、拟杆菌门(蓝色)、放线菌门(紫色)、变形菌门(绿色)和梭杆菌门(灰色)的 38 个代表性簇的泛基因组拟合曲线。泛基因组大小是从每个簇中包含的所有菌株组合中累积的。
我们还探索了肠道细菌泛基因组中参与
丁酸合成和抗生素耐药性的基因分布
。功能注释显示六个簇包含完整的乙酰辅酶 A 到丁酸的生物合成途径(图 4a)。其中,
F. prausnitzii、E. rectale、产丁酸盐细菌SS3_4和Roseburia sp
。CAG:45 在其核心基因组中包含完整的通路,表明这些物种中产生丁酸盐的功能是高度保守的。该结果与报道的这些物种的丁酸盐生产能力一致。为了探索已建立的泛基因组中抗生素抗性的分布,我们在每个泛基因组中注释抗生素抗性基因 (ARG)。与之前的报告一致,
四环素抗性基因广泛存在于这些簇的可有可无的基因组中
(图 4b)。值得注意的是,
大肠杆菌在其泛基因组中几乎包含所有 ARG
(25 个中的 23 个),其中一半存在于核心基因组中(图 4b)。相比之下,双歧杆菌属物种,包括双歧双歧杆菌、青春双歧杆菌、长双歧杆菌和假链状双歧杆菌,其泛基因组中很少包含 ARG。
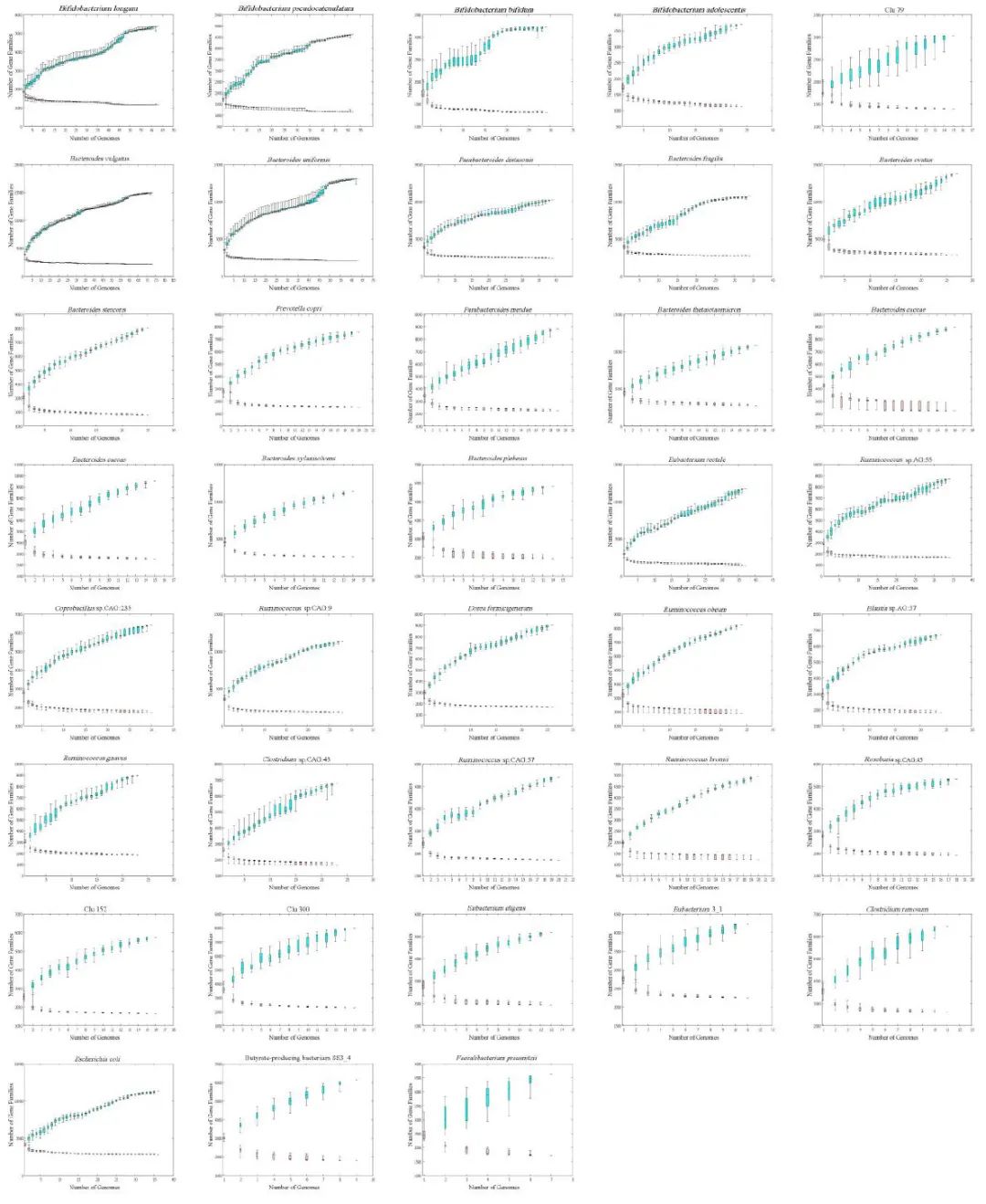
附图9. 38 个簇的泛基因组和核心基因组分析
泛(青色)和核心(粉红色)基因组中基因家族的数量被绘制为 38 个簇的基因组数量的函数。箱线图表示第 25 个和第 75 个百分位数,中位数显示为水平线,胡须设置在第 10 个和第 90 个百分位数

图 4:38个代表簇的泛基因组分析
Fig. 4: Pan-genome analysis of 38 representative clusters.
a,参与丁酸生物合成途径的基因在核心基因组(粉红色)和可有可无的基因组(青色)中的分布。下面显示了从乙酰辅酶 A 生物合成丁酸的两条途径。在核心基因组和泛基因组中具有完整丁酸生物合成途径的物种分别以粉红色和青色突出显示。Thl,硫解酶;Hdb, β-羟基丁酰-CoA 脱氢酶;Cro,巴豆酶;Bcd,丁酰辅酶A脱氢酶(包括电子转移蛋白α和β亚基);但是,丁酰辅酶A:乙酸辅酶A转移酶;Ptb,磷酸丁酰转移酶;Buk,丁酸激酶。b,ARGs 在核心基因组(粉红色)和可有可无的基因组(青色)中的分布。
为了更好地了解细菌功能在核心和可有可无的基因组中的分布,我们使用直系同源群 (COG) 数据库对基因组进行了注释。这表明核心基因组中的几种管家功能显着丰富,包括翻译后修饰、蛋白质周转和分子伴侣(O,P = 7.28 × 10
-12
);翻译、核糖体结构和生物发生(J,P = 7.28 × 10
-12
);能源生产和转化(C,P = 7.28 × 10
-12
);氨基酸转运和代谢(E,P = 7.28 × 10
-12
);核苷酸转运和代谢(F,P = 7.28 × 10
-12
);辅酶转运和代谢(H,P = 1.46 × 10
-11
);脂质转运和代谢(I,P = 2.40 × 10
-10
);和无机离子转运和代谢(P,P = 2.40 × 10
-10
)(补充图10)。相比之下,在可有可无的基因组中丰富的 COG 类别包括细胞壁 - 膜 - 包膜生物发生(M,P = 2.70 × 10
-9
);细胞运动性(N,P = 3.11 × 10
-5
);信号转导机制(T,P = 0.00039);细胞内运输分泌和囊泡运输(U,P = 1.22 × 10
-7
);防御机制(V,P = 7.28 × 10
-12
);转录(K,P = 3.64 × 10
-11
);复制重组和修复(L,P = 7.28 × 10
-12
);和功能未知(S,P = 0.03111)。其余的 COG 类别在核心和可有可无的基因组中没有显着差异。
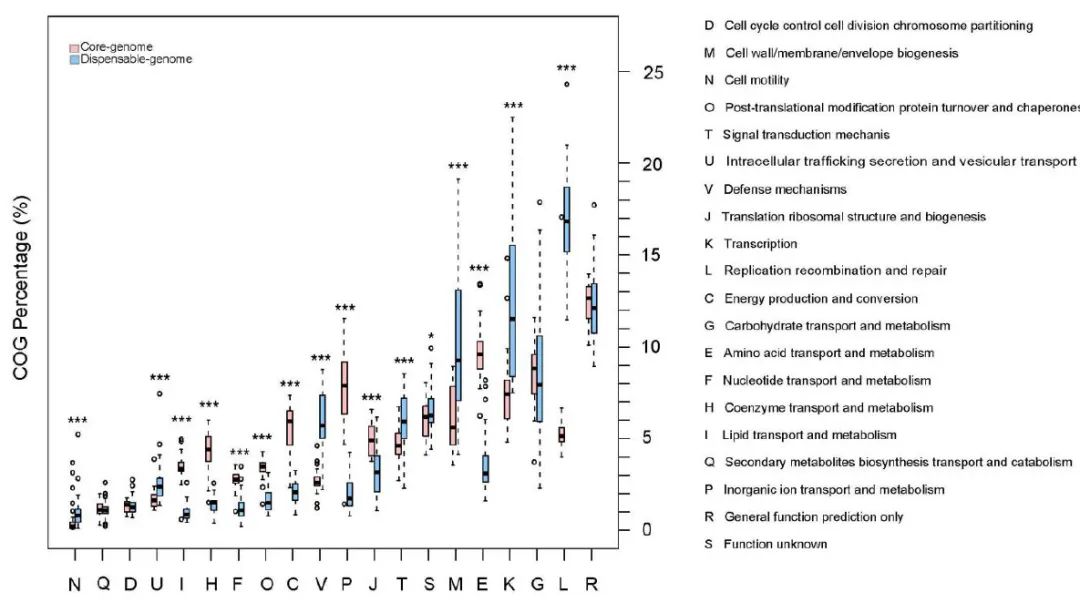
附图10. 核心基因组和可有可无的基因组中的 COG 分布
将核心基因组(粉红色)中 20 个 COG 的百分比与 38 个簇的泛基因组(青色)中的百分比进行比较。改善的显着性由两侧 Wilcoxon 秩和检验确定(
,P < 0.05;
,P < 0.01;
,P < 0.001)。D 的精确 P 值为 0.931,M 为 2.70×10-9,N 为 3.11×10
-5
,O 为 7.28×10
-12
,T 为 3.88×10
-4
,U 为 1.22×10
-7
,7.28×10
-12
为 V,J 7.28×10
-12
,K 3.64×10
-11
,L 7.28×10
-12
,C 7.28×10-12,G 0.261,E 7.28×10
-12
,7.28×10- F 为 12,H 为 1.46×10
-11
,I 为 2.40×10-10,Q 为 0.874,P 为 2.40×10
-10
,R 为 0.365,S 为 0.031。每个箱线图说明估计的中位数(中心线) )、上下四分位数(框限)、1.5 × 四分位距(须线)和 COG 百分比的异常值(点)
讨论
我们使用了 11 种培养条件来分离肠道细菌,并建立了 6,000 多个分离株的菌库。从这些分离株集合中,我们生成了 1,520 个高质量的参考基因组草图。由此产生的 CGR 在属和种水平(包括低丰度物种)的高覆盖率证明了基于培养的方法从肠道微生物群中分离菌株的价值。与此一致的是,在最近的两项研究中,已经成功培养了大量以前被认为无法培养的肠道细菌物种。尽管 CGR 菌库的新物种与这两项研究之间存在一些重叠,但 CGR 包含 659 个额外的基因组(代表 209 个集群或物种)。我们的培养方法可用于扩大 CGR,直到它被可培养肠道细菌的基因组饱和。之后,单细胞测序可用于研究不可培养细菌的基因组,总体目标是定义一组饱和的肠道微生物群参考基因组,以支持更好地了解肠道微生物组生物学。
我们应用了 CGR 基因组数据集来为肠道细菌分配功能。例如,我们发现毒力因子和抗生素抗性基因在克雷伯菌属、埃希氏菌属、柠檬酸杆菌属和肠杆菌属中富集,它们是临床样本中经常分离的机会性病原体。这些细菌中丰富的信号转导和细胞运动基因可能进一步促进其致病性。值得注意的是,变形菌还具有丰富的异生物质降解基因,这可能会影响药物治疗中患者的药物代谢。与此一致的是,最近的一项研究报告称,瘤内变形菌可以将化疗药物代谢成无活性形式,从而减弱癌症治疗的疗效。参与有益功能(如
聚糖降解和维生素 B 合成)的基因在拟杆菌属中富集,与其在人类肠道中的互利作用一致
。值得注意的是,我们发现
拟杆菌属物种包含大量参与鞘脂和类固醇激素合成的基因,这表明它们具有调节哺乳动物细胞信号传导的潜力
。为了支持这一点,最近的一项研究报告称,脆弱拟杆菌可以利用鞘脂信号传导在肠道中实现共生。值得注意的是,参与聚糖降解和鞘脂代谢的基因也在双歧杆菌属(另一种众所周知的肠道共生微生物)中富集。然而,参与这两种途径的基因在普氏菌属中并不丰富,这表明与拟杆菌门的其他成员相比,普氏菌具有独特的功能。这可能解释了肠道微生物群中普氏菌和拟杆菌的相对丰度之间观察到的负相关。肠道细菌在雌激素代谢中的潜在作用早已得到认可 ,但仍缺乏详细的机制研究。探索肠道细菌的这种独特功能在激素相关健康或疾病中的意义将会很有趣。CGR 还能够鉴定出几种潜在的产细菌素菌株,值得进一步验证。
CGR 将改进宏基因组分析、基因组变异分析、功能表征和泛基因组分析。分离出的肠道细菌菌株已保存在中国国家基因库 (CNGB) 中,可用于旨在改变微生物群功能的研究、作为新型益生菌或用于验证疾病相关细菌标志物。
方法 Methods
粪菌厌氧培养
粪便样本是从 155 名在采样前最后一个月未服用任何药物的健康人类捐献者身上收集的。补充表 2 中给出了详细信息。将样品立即转移到厌氧室(Bactron Anaerobic Chamber,Bactron IV-2,Shellab,USA),在补充有 0.1% 半胱氨酸的预还原磷酸盐缓冲盐水(PBS)中匀浆,然后稀释并铺展在具有不同生长培养基的琼脂平板上(补充表 1)。平板在厌氧条件下在 90% N2、5% CO2 和 5% H2 的气氛中在 37°C 下培养 2-3 天。挑取单菌落并在新平板上划线以获得单克隆。所有菌株均在 –80 °C 下储存在含有 0.1% 半胱氨酸的甘油悬浮液 (20%, v/v) 中。155 个样本的收集得到了 BGI 生物伦理和生物安全机构审查委员会的批准,编号为 BGI-IRB17005-T1。所有协议均符合赫尔辛基宣言,并获得了所有参与者的明确知情同意。CGR(可培养基因组参考)中的细菌存放在中国国家基因库(http://ebiobank.cngb.org/index.php?g=Content&m=Hql&a=sample_5&id=393 )。
全基因组测序和从头组装
DNA提取
将培养至固定相的分离物在 4°C 下以 7,227g 离心 10 分钟,并将所得沉淀物重新悬浮在 1 ml Tris-EDTA 中。对于细菌细胞裂解,加入50 μl 10% SDS和10 μl蛋白酶K(20 mg/ml),溶液在55℃水浴中孵育2 h。使用苯酚-氯仿法提取释放的基因组 DNA。
基因组测序和组装
构建插入片段大小为500 bp的双端文库,并在Illumina Hiseq 2000平台上测序,获得每个样本约100 × clean数据。使用 SOAPdenovo 2.0441 组装读取以形成支架,通过
RNAMMer 1.242 从中提取 rRNA 基因
。内部流程用于获得包含正交设计的最佳装配,以研究 L,M,d,D,L,u,G(SOAPdenovo 的参数)和单因素设计以研究 K(SOAPdenovo 的参数)通过综合考虑重叠群 平均长度、最长支架和 rRNA 分数。在ionProton平台上构建并测序了插入大小为240 bp的文库,该平台为每个样品产生了约100 × 干净的数据。通过
SPAdes(版本 3.1.0) 组装读取以形成支架
。
基因组质量评估
采用人类微生物组计划 (HMP)和 rRNA(5 s、16 s 和 23 s)完整性的六个高质量草案组装标准来确保组装质量。标准是 (i)
90% 的基因组组装必须包含在 contigs 中 > 500 bp
,(ii)
90% 的组装碱基必须位于 > 5 × read 覆盖率
,(iii)
contig N50 必须 > 5 kb
,(iv)
支架 N50 必须 > 20 kb
,(v)
平均重叠群长度必须 > 5 kb
,并且(vi) >
90%的核心基因必须存在于组装中
。
多基因组分离物的拆分
分离物中的多基因组最初使用 CheckM(污染 > 5%)进行鉴定,并通过手动检查 G + C 百分比与测序深度的关系图进行确认。开发了一个内部管道来将多基因组的支架分成单个基因组。简而言之,首先使用 R 的 dbscan 函数(包“fpc”)根据 G + C 百分比与测序深度值对多基因组中的支架进行拆分。使用 CheckM 检查分裂基因组的“完整”和“污染”。对于“完整” > 100% 或“污染” > 15% 的分裂基因组,使用额外的物种指定管道来获得它们最接近的参考(ANI 值 > 90%)。最后,使用BLASTn(-e 1e-5 -FF -m 8,blastn hits’length > 90 nt,query scaffold coverage ≥ 50%)将每个分裂基因组中的错误分裂支架映射回最近的参考基因组,以获得最后分裂的基因组。
海量种属分配过程
NCBI 检索的原核基因组
NCBI ftp 站点上的所有完整基因组(更新时间为 2014 年 11 月 19 日)和基因组草图(更新时间为 2014 年 8 月 8 日)都下载到本地服务器。具有多个 NCBI 分类标识符 (taxid) 或基因组序列不可用或非原核来源的项目被删除,而冗余项目仅保留一个。结果,获得了 24,552 个基因组、19,116 个基因组规模的氨基酸序列及其分类信息。
物种水平分类分配的平均核苷酸同一性 (ANI)
每个查询基因组的分类分配由所有 NCBI 可用的原核基因组的分类信息确定。获得了所有基因组和每个查询基因组的四碱基特征图谱。在每个查询基因组和所有基因组之间执行 Pearson 相关性测试,产生一个参考列表,按每个查询基因组的相关系数递减排序。然后根据参考列表将查询基因组与参考基因组一一进行ANI成对比对(tetra-base profile的Pearson相关检验:相关系数 > 0且P < 0.001),直到ANI值大于95%进入前500名项目(在本例中定义为已分配)或参考项目编号超过 500 且没有任何 ANI 值大于 95%(在本例中定义为未分配)。
用于属级分类分配的保守蛋白 (POCP)的百分比
每个查询基因组的分类分配由所有 NCBI 可用的原核基因组的分类信息确定。获得所有参考基因组和未基于ANI进行物种分配的查询基因组的四碱基特征图谱。在每个查询基因组和所有参考基因组之间执行 Pearson 相关性测试,产生一个参考列表,按每个查询基因组的相关系数递减排序。然后根据参考列表在查询基因组和参考基因组之间一一进行POCP计算,直到前500个项目中的POCP值大于50%(在这种情况下定义为“分配”)或参考项目数量超过500个没有任何大于 50% 的 POCP 值(在这种情况下定义为“未分配”)。
16S rRNA 序列分析和新物种确定
除了16个基因组提取失败外,使用R
NAmmer从分离株基因组中提取了16 S rRNA基因序列。序列在 EzBioCloud (http://www.ezbiocloud.net) 中进行质量控制处理。物种级操作分类单位 (OTU) 使用 mothur50 进行分类,同一性为 98.7% 作为物种级临界值,94.5% 和 86.5% 的临界值分别用于属和科 51
。
CGR 与来自其他研究的基因组数据集的比较
为了将 CGR 中存档的新基因组和新物种与最近两项研究中发现的进行比较,我们下载了 Browne 等人报告的 215 个基因组和 Lagier 等人报告的 169 个基因组。通过用这些新下载的基因组替换 NCBI 参考,我们采用了如上所述的类似 ANI 管道进行物种级比较。如果我们的 1,520 个高质量基因组中的查询基因组与任何一个参考基因组(四碱基谱的 Pearson 相关检验:相关系数 > 0 和 P < 0.001)之间的成对 ANI 值大于 95%,则定义“可比对” ;如果不是,则该物种被定义为“未比对”。
物种簇的构建
在 1,520 个高质量基因组中进行成对 ANI 比对,然后
使用 R 包中的 hclust 进行距离为 0.05(相当于 95% ANI)的层次聚类
。
一组 40 个普遍保守的单拷贝基因在细菌和古细菌中编码蛋白质,用于构建系统发育树
。使用 specI 和 prank 检测和比对标记基因。对齐由trimal 修剪并与内部脚本连接。使用最大似然法和 RAxML(版本 8.2.8)推断出一个系统发育树,用于以硒化根瘤菌 ATCC BAA 1503 (taxoid:1336235) 作为外群的簇的代表性基因组(簇中最长的 N50),并在iTOL (http://itol.embl.de/)在线。
基因组功能注释
对 1,520 个高质量基因组进行了功能注释。使用 Genemark 鉴定基因。编码基因的翻译氨基酸序列与 RAPSearch (-sf -e
1e-2
-v 100 -u 2) 对照京都基因和基因组百科全书(KEGG 76 版,
查询匹配长度高于50%
) 或使用 BLASTp (-e 1e-2 -FT -b 100 -K 1 -a 1 -m 8) 对抗抗生素抗性基因数据库 (ARDB)(查询和主题
匹配长度均高于 40%
,具有相似性)高于ARDB推荐的阈值)、毒力因子数据库(VFDB)(
查询匹配长度高于50%,相似性高于60%
)和细菌素数据库(从BAGEL3下载,相似性高于60%)。针对综合抗生素耐药性数据库 (CARD) 的基因注释是使用耐药基因标识符进行的,该标识符可作为 CARD 网站下载部分中的可下载命令行工具使用默认参数进行。
宏基因组样本的比对率
首先使用 SOAP2(默认参数,除了 -m 100 -x 1000 -r 1 -l 30 -v 5 -c 0.95 -u)将宏基因组读数与 IGCR 的参考基因组(来自 IGC6 的 3,449 个测序原核基因组)进行比对。然后
将未比对的读数与新测序的 CGR 基因组进行比对
。计算不同样品的read mapping比,样品间的
差异通过R中的Wilcoxon检验确定
。
基因和蛋白质多样性分析
基因簇
使用 CD-HIT 和默认参数对 1,759 个 CGR 基因组中的 5,749,641 个基因和 3,449 个 IGCR 基因组中的 11,330,042 个基因进行聚类,参数 -G 0 -aS 0.9 -c 0.95 -M 0 -d 0 -r 1,相当于 -g 1对于较短的序列,95% 的局部序列同一性超过 90% 的比对覆盖率。一个簇由两个或多个基因组成。根据样本名称字母顺序绘制基因簇累积曲线,前部为IGCR,后部为CGR。
蛋白质簇
从 1,759 个 CGR 基因组中的基因翻译的 5,749,641 个蛋白质序列和从 3,449 个 IGCR 基因组中的基因翻译的 11,330,042 个蛋白质序列使
用 kClust 算法 使用默认参数进行聚类,这相当于 20-30% 最大成对序列同一性超过 80%
比对长度与簇的最长序列或种子。一个簇由两个或多个蛋白质序列组成。根据样品名称字母顺序绘制蛋白质簇累积曲线,前部为IGCR,后部为CGR。
SNP 识别和相似度评分
使用 SOAP2 将来自 CGR 的 1,520 个基因组与来自 250 个 TwinsUK 样本的测序读数进行比对,一致性≥90%。用于 SNP 分析的代表性基因组是根据先前描述的三个标准确定的。得到的 282 个满足这些标准的基因组(补充表 7)被用作使用 SAMtools 进行 SNP 调用的参考(
频率 > 1% 并由≥4 个读长支持
)。将先前研究中使用的参考基因组(152 个基因组)与本研究的 CGR(282 个基因组)中使用的参考基因组进行比较,以使用 ANI ≥ 95% 作为阈值(物种水平)来识别共享的和新的参考基因组。
38个簇的泛基因组分析
使用
细菌泛基因组分析工具 (BPGA)
将包含十多个基因组(来自 CGR 和 NCBI)以及 Fecalibacterium prausnitzii(七个基因组)和产丁酸细菌 SS3_4(九个基因组)的簇用于泛基因组分析管道。
簇中所有成员共享的一组基因被定义为核心基因,而成员中部分共享的基因(附属基因)和簇中单个成员所独有的基因(独特基因)被定义为可有可无的基因
。38 个簇的泛基因组拟合曲线由 BPGA 工作流程生成并绘制在 R (v.3.3.3) 中。KEGG 和ARDB 对38 个簇的泛基因组中基因的功能进行了注释,使用的参数与用于基因组功能注释的参数相同。乙酰辅酶A到丁酸的生物合成途径是根据先前的研究产生的,并且根据NCBI蛋白质数据库的功能注释和BLAST鉴定相关酶(截止
1e-5,同一性≥70%,覆盖率≥70%
)。COG 数据库还用于通过 BPGA 管道识别核心部分和可有可无的部分中的功能分布。使用在 R (v.3.3.3) 中实施的 Wilcoxon 检验检查了核心基因组和可有可无的基因组中 COG 分布之间差异的显着性。
数据可用
1,520 个 CGR 菌株的组装基因组草图和注释信息以登录号 PRJNA482748 (
可作为细菌基因组提交的参考模板,包括核酸、基因组和蛋白序列,样本和和组装信息
) 保存在 NCBI,这些数据也可在中国国家基因库 (CNGB) 核苷酸序列档案 (CNSA;登录号 CNP0000126) 中找到。CGR 中的所有菌株都保存在 CNGB 中,CNGB 是中国的一个非营利性公共服务机构。每个菌株的登录代码在补充表 5 (Genebank_id) 中给出。研究人员可以通过 http://ebiobank.cngb.org/index.php?g=Content&m=Hql&a=sample_5&id=393# 探索菌株信息并请求菌株。
参考资料
Yuanqiang Zou, Wenbin Xue, Guangwen Luo, Ziqing Deng, Panpan Qin, Ruijin Guo, Haipeng Sun, Yan Xia, Suisha Liang, Ying Dai, Daiwei Wan, Rongrong Jiang, Lili Su, Qiang Feng, Zhuye Jie, Tongkun Guo, Zhongkui Xia, Chuan Liu, Jinghong Yu, Yuxiang Lin, Shanmei Tang, Guicheng Huo, Xun Xu, Yong Hou, Xin Liu, Jian Wang, Huanming Yang, Karsten Kristiansen, Junhua Li, Huijue Jia, Liang Xiao. (2019). 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nature Biotechnology 37 179-185 doi: 10.1038/s41587-018-0008-8
高颜值免费 SCI 在线绘图
(
点击图片直达
)
最全植物基因组数据库IMP
(
点击图片直达
)
往期精品
(
点击图片直达文字对应教程
)
机器学习