在绝大数药物靶点中酶学靶点是一个大类。本文主要以酶催化的基本化学反应原理为起点,概述和阐述下酶催化反应的几个基本概念Km,Kcat和Ki,以及其测定方法,分析下一个灵敏的酶学筛选方法开发的几个关键步骤,概括论述下酶抑制剂的类型以及在实验检测过程中不同类型酶抑制剂的IC50和Ki之间的关联。
酶是生命体内化学反应的催化剂,能够在生理温度和常压下加快化学反应的速度,几乎所有的细胞活动进程都需要酶的参与以提高效率。特定酶活性或者表达量的增强或者缺少都有可能导致生命活动的异常,疾病的发生。在药物研发过程中酶是十分重要的靶点,也是十分有潜力和效果的靶点,如最近在中国上市的吉利德的慢性丙型肝炎新药索华迪(索磷布韦),其效果和价格一样让人印象深刻,一个疗程6万人民币左右,对于基因1,2,3,6型HCV具有抗病毒活性,治愈率高达92%-100%,其作用的靶点是NS5B RNA聚合酶;又如晚期大肠癌的一线用药是伊立替康,其代谢活性成分SN-38是DNA拓扑异构酶Ⅰ抑制剂,其与拓扑异构酶Ⅰ及DNA形成的复合物能引起DNA单链断裂,阻止DNA复制及抑制RNA合成,为细胞周期S期特异性。首款获得FDA批准的双药HIV疗法新药Juluca,其两个活性成分,Dolutegravir是HIV-1整合酶链转移抑制剂(integrase strand transfer inhibitor,INSTI);Rilpivirine是一款非核苷逆转录酶抑制剂(non-nucleoside reverse transcriptase inhibitor,NNRTI)。酶作为一个很有潜力的靶点,如何建立一个简单高通量的酶学抑制物的筛选方法是这篇文章笔者主要讨论的问题。我们可以重温一下酶学基本的反应原理开始
绝大多数酶学反应都可以用下图简单的模型进行描述:
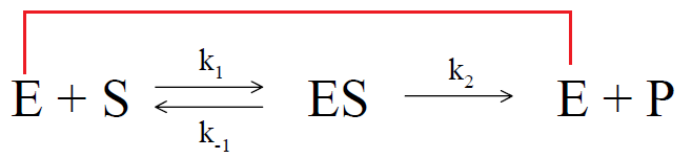
E代表酶,S代表底物,P代表催化产物。ES代表在催化反应开始前形成的中间态产物酶-底物复合物,E和S形成ES复合物的反应是可逆反应的过程,k
1
是E和S形成ES复合物的结合速率常数,相当于配体受体结合时的K
on
或者K
a
, k
-
1
是ES解离形成E和S的解离速率常数,相当于配体受体结合时的K
off
或者K
d
(关于以上概念可参考笔者前期的文章--
生物功能学筛选评价技术之亲和力评价篇
);ES解离形成E和P的过程在反应初期一般认为是不可逆的过程,反应产物E会被反应体系中的过量的S捕捉,k
2
是ES形成E和P的速率常数,也被称之为K
cat
,相当于配体受体结合时的K
on
或者K
a
酶催化效率:
一个酶相对于其底物的催化效率主要由两个方面决定K
cat
/K
m
, K
m
值(米氏常数)越小, K
cat
值越大,催化效率越高。
K
m
值的化学本质是E和S可逆反应过程中的解离平衡常数K
D
,
反映的是E和S亲和力的强弱,K
m
值越小亲和力越强,效率越高。K
cat
值是ES解离形成E和P的速率常数。
K
m
值和K
cat
值测定
米氏方程描述了在稳态反应条件下酶催化反应中初始反应速度V
0
和底物浓度 [S]的相关关系:
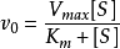
当V
0
等于1/2的 V
max
时,底物浓度 [S]为K
m
值。
在实际的检测过程主要包括以下几个步骤:
1. 少量固定浓度的酶(nM或者pM级别),梯度浓度稀释的底物(底物浓度相对于酶浓度由远远过量,mM到μM)进行分组,酶催化反应;
2. 每个分组对酶催化反应的产物进行动力学检测(酶标仪进行荧光,吸光度检测或者HPLC检测),选取各分组信号时间的线性反应区域进行V
0
计算;
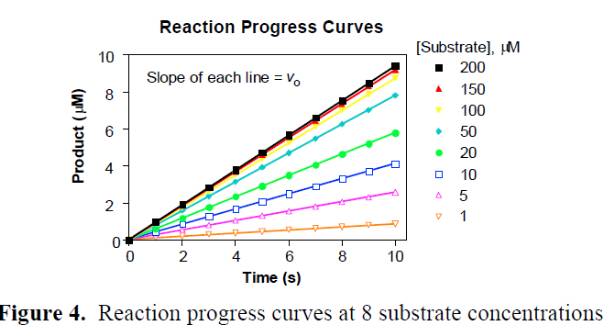
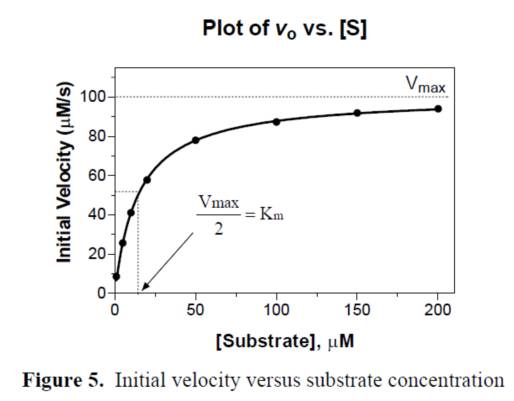
3. 选取梯度浓度稀释的底物动力学检测的线性区域的斜率作为初始反应速度V
0
进行数据米氏方程拟合,拟合图形的平台期为V
max
, 1/2的V
max
时V
0
对应的底物浓度值是Km值。处理结果示意图如下:
4. 当酶催化反应达到最大速度时,S远远大于E,所以所有的E均被S捕获,E和S的逆反应速度可忽略不计,[ES]=[E]
V
max
=[ES]*K
cat
=[E] *K
cat
,所以
K
cat
= V
max
/[E]
以上是Km和Kcat的测定和计算过程,值得注意的是在实际的实验过程中需要注意以下几点:
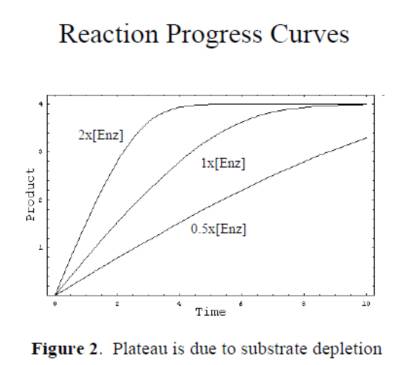
1. 必须是动力学读数,选取初始反映的线性段的斜率作为初始反应的速度。终点法读数的结果很多时候并不可靠,应为酶催化反应随着时间的进行底物的消耗,反应速度会从匀速到非均速变化,如果采用终点法可能得到不真实的初始速度,如下图:
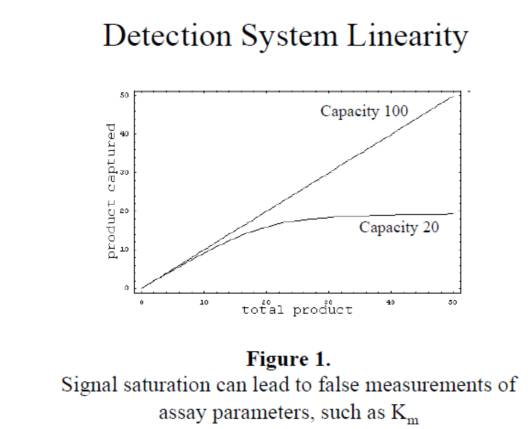
2. 必须确认仪器的检测线性,当酶催化反应,产物的量过多时,超过仪器检测的线性区域时,检测的产物的量不准,影响到后续参数的计算,如下图:
酶学筛选方法的几个核心参数包括酶,反应环境,底物和抑制物。一般来讲反应环境需要查阅文献进行调研,确定PH值,离子强度,洗涤剂等对酶活的影响,底物可以根据酶催化原理选择天然底物,也可以选择合成底物(底物的选择和后续配套的检测方法紧密相关)。一个酶学方法的开发可能涉及到以下一些因素:
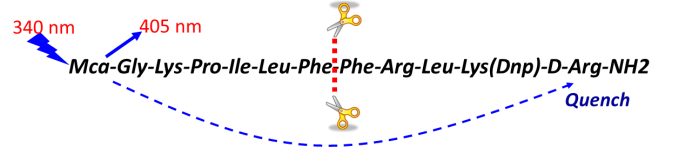
底物的选择和配套的检测方式,如下图某蛋白水解酶,其识别的位点是氨基酸线性表位,可以人工合成多肽,在其N端和C端分别加入荧光基团和淬灭基团,正常状态下或者酶活抑制状态,该多肽在酶标仪340nm激发时,其发射光会被淬灭基团吸收,在405nm无信号产生;酶催化反应会将该多肽剪切,当酶标仪340nm激发时,405nm有信号产生。
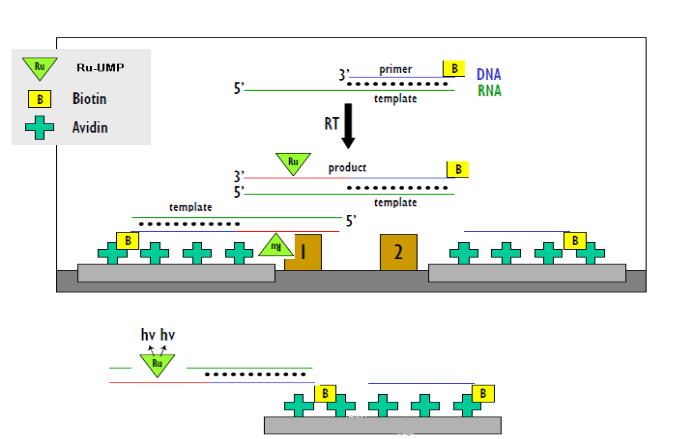
又如某病毒逆转录酶,合成RNA模板和对应引物,在其引物上进行生物素标记,其中碱基上进行Ru标记,随着酶催化的逆转录反应的进行,引物上不断有Ru标记碱基上去形成核苷酸序列,通过亲和素的板子捕捉引物,检测Ru信号值来评判酶的催化和抑制的活性。
酶,底物浓度的选择和反应时间的选择
当酶对应的底物确定以后,选取少量的酶浓度(pM或者nM)对底物进行Km测定,选取底物的Km浓度作为筛选的反应浓度,选取该浓度下线性的反应时间内的某个时间点作为反应终止或者检测的时间。酶和梯度稀释的化合物进行预孵育后,加入Km浓度的底物,反应一段时间后进行检测,这个就是一般酶学筛选模型。
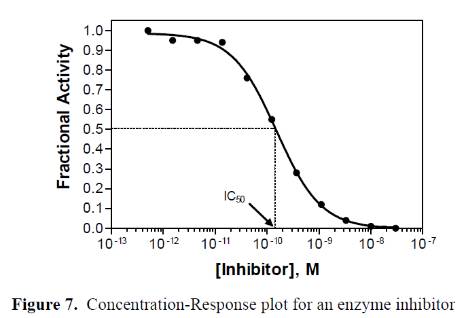
对于靶点酶的抑制剂,主要可以归纳为三类(competitive竞争性, noncompetitive非竞争性, uncompetitive不竞争性)
competitive竞争性抑制剂只和自由的酶结合,通常情况下和底物共同竞争结合位点(活性中心),当然也有竞争性抑制剂和底物互斥的情况出现,两个中只有一个能够和自由的酶结合。在竞争性抑制剂存在的情况下,酶相对于底物的Km和Vmax测定时,测得得表观Km值增加,Vmax值不变,如下图所示,几种竞争性抑制剂的图示:





