请注意,这里
CAPs
不是很多帽子,也不是什么共同农业政策
(Common Agricultural Policy)
,而是
Cleaved AmplifiedPolymorphic Sequences
。
随着越来越多的测序数据公布,尤其是海量重测序数据的释放,很多物种中的基因组多态性位点尤其是
SNP
,不能说完全被发现,但是接近饱和是真的。这几年国内测序环境实在是太好了
(钱多人不傻,不要跟我说是因为情怀),热的不能再热,测几百个上千个种,做个群体结构、重要性状
GWAS
、驯化分析,堆堆数据,自圆其说,重要物种花钱花的多的,发个
NG
。影响力小的物种发个
NC
。文章一出,鸡犬升天。。。数据随之束之高阁(有点儿夸张,但也差不多
^_^
)。其实内心是很嫉妒的有没有,为什么人家资源那么多还那么有才(
qian
)。

其实花巨资测的这些数据,除了发一个大文章来提高影响力以外,其他的也很有价值。如何利用这些种质资源,结合分子标记进行分子标记辅助育种
(
MAS, GS
),提高人们生活质量这才是研究的最根本目的吧。至于什么进化啊,目前没看出来什么应用价值吧,请同行们拍砖,洗耳恭听

情
怀说
完了,
说说为
什么要做
这
个高通量
CAPS marker
设计吧
。
官方是这样解释的:
1.
相比
Indel
来
说
,基因
组
范
围
内
SNP
的数量是其
10
倍不止,因
为设计
marker
比
较
麻
烦
也有限制,除了高通量
测
序,芯片,
这
么多
资
源很少被利用,
虽
然
现
在也有什么
fludigum
。身
为
科研人
觉
得
这样
浪
费资
源太可耻了。
其实是这样的
:
1.
SSR
已
经
被人
设计
完了。
2.
Indel
太
简单
,
显
示不出能力来啊。会
CAPS
还能设计不出来
Indel
么。
3.
发个
文章
耍耍,作为数据库类型的文章,可能引用率还是很高的。你看到的没错,是
文章
,可以发
文章
!!!看到这里是不是心动了,没看到这就关闭的同学那就不怪我咯
。
进入正题
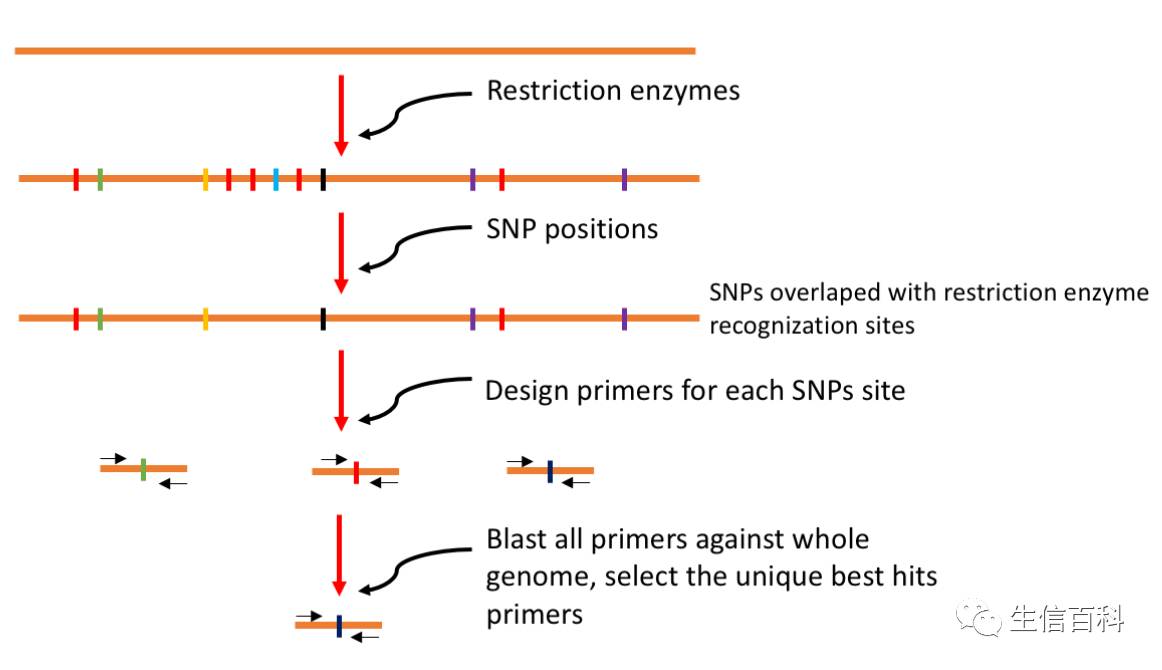
基本思路如下图:
1.
用所有可用的限制性内切
酶对
要研究物种的全基因
组进
行
酶
切,
统计酶
切位点。
2.
将目前公布的
SNP
数据与
酶
切位点
进
行比
对
,提取重合位点。(
这
些位点就可以
进
行
CAPS
标记开发
)
3.
对这些位点上下进行
150bp
的片段
进
行高通量引物
设计
。跑
过
胶的同学都知道,
对
marker
来
说
,
200bp
以内
还
是很好用的
^_^
。省
酶
省
时间
啊(就是
这
么
抠
)。
4.
对你的
primer
进行物种内
blast
,如果你的引物和基因
组
其他位置高度匹配,那么
这
种引物就要舍弃。
5.
实验验证。你设计了这么多引物,总得验证下效率吧,不然谁敢用啊
。
6.
做个小界面程序方便
别
人利用,有数据
库
的放到网站上最好哦。
7.
写个文章吧。(文章大小取决于你研究物种的影响力哦
……^ ^
)
准备数据:
1.
研究物种的基因
组
,
fasta format
。
2.
研究物种已公布的
SNP
数据,要有位点信息和
allele
信息。
所需工具:
1.
EMBOSS
软件。请自行查找安装,可能有点儿小麻烦,主要是限制性内切酶的数据库问题,这里不讨论如何安装,如果遇到麻烦请留言,留言多了的话再加一期如何安装这个软件
。
2. Primer3, 设计引物用。
2.
python
,
没有
python
我
该
如何
过滤
我的数据。。。
perl or any other you want^^.
3.
R,
并不是所有
语
言都能像
R
这么无脑使用的
^ ^
。
先抛个话题,下期我们正式搞起。