RNA-seq新赛季,DEG分析流程迎来新包(一大波旧包喜迎退环境)
写在前面
惊闻Tidybulk发在了Genome Biology。一群人想破头都明白这么多seq相关包和在线工具,凭什么tidybulk能发的那么高。难道就因为你叫泰迪?跑了一遍流程后,感觉这个包还是有点意思。流程奉上,独自码不如一起码。
本文参考tidybulk官方教程GitHub和2020年Workshop。
首先读取需要的包。这里推荐直接从github安装tidybulk。从BiocManager上安装的是老版本。
#tidybulk最新版
#devtools::install_github("stemangiola/tidybulk")
#devtools::install_github("stemangiola/tidyHeatmap")
library(tidyverse)
library(tidybulk)
library(tidyHeatmap)
library(ggrepel)
library(corrplot)
library(ggforce)
library(aplot)
library(GGally)
这次跑点不是人的数据集,https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155319因为作者直接上传了表达矩阵,我们这边直接从NCBI读取需要的文件。具体视自己的网速,网速不给力请用意志克服。
#清空
rm(list = ls())
#读取文件
expr_matrix <- read_delim("https://ftp.ncbi.nlm.nih.gov/geo/series/GSE155nnn/GSE155319/suppl/GSE155319_genes_fpkm_expression.txt.gz", delim = "\t")
glimpse(expr_matrix)
#简单的从文件里提取需要的列并简化列名
count_matrix <- expr_matrix %>%
select(transcript_id, dplyr::starts_with("count"))
names(count_matrix) <-
names(count_matrix) %>%
str_remove("count.") %>%
str_remove("14")
count_matrix %>% glimpse()
考虑到永远有新人入坑,这里简单介绍一下tidy系列包的特点。
tidy包会使用通道符(%>%)连接命令,使用tibble格式的数据框架以及使用ggplot图形语法进行可视化。严格来讲tidyHeatmap因为无法跟ggplot兼容(底层使用Gu大的complexheatmap),所以含泰(tidy)度不足。
至于%>%连接符的作用,一张图简单介绍一下:
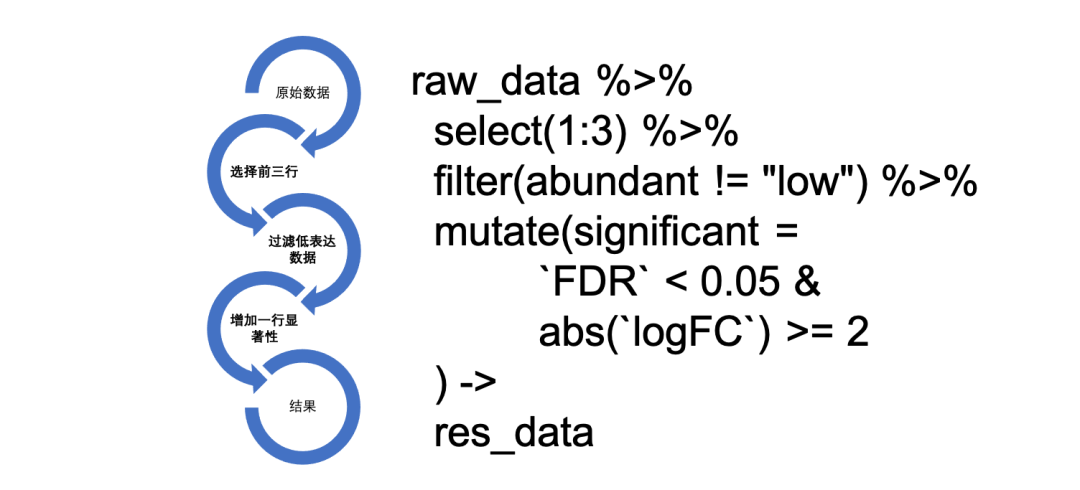
左边是代码运行的流程,右边是代码。代码的可读性很强,写起来也很爽……
像极了我小时候一句话能写一个自然段……
正式流程
跑一个正常的RNA-seq流程我们首先要得到
三张图
(aka生物技能树ISO):相关系数矩阵,高差异表达基因热图和PCA聚类图。tidybulk的工作流程里还多了
基因表达分布图
。
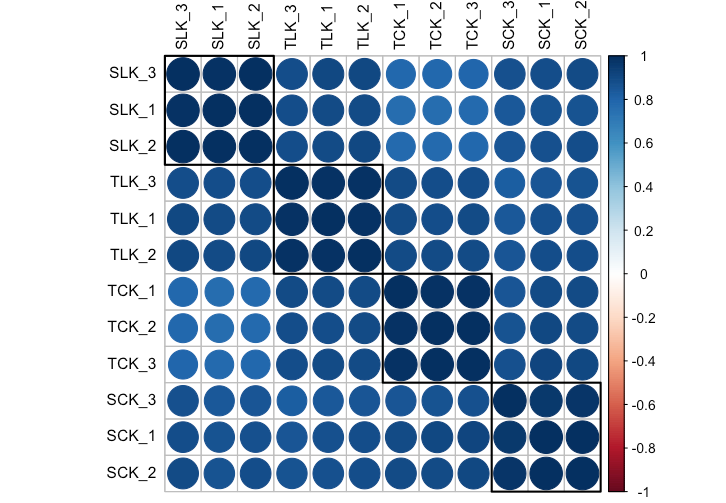
相关系数矩阵
首先是获得各个重复和处理的相关系数矩阵
count_cor <- cor(count_matrix[-1])
count_cor
相关系数矩阵这里介绍两种作图方法:
首先是比较经典的corrplot
num <- colnames(count_cor) %>%
str_remove("_[0-9]") %>%
unique()
corrplot(count_cor,
order = "hclust",
addrect = length(num),
tl.cex = .9, tl.col = "black")
另一种是使用tidyheatmap的相关系数矩阵
count_cor %>%
as_tibble(rownames = "name") %>%
pivot_longer(!name, "sample", "cor") %>%
mutate(condition = gsub("\\_\\w","\\1",name),
Condition = gsub("\\_\\w","\\1",sample)) %>%
heatmap(
name,
sample,
value,
palette_value = circlize::colorRamp2(c(-1, -.5, 0, .5, 1), viridis::viridis(5)),
rect_gp = grid::gpar(col = "white", lwd = .5)
) %>%
add_tile(Condition)
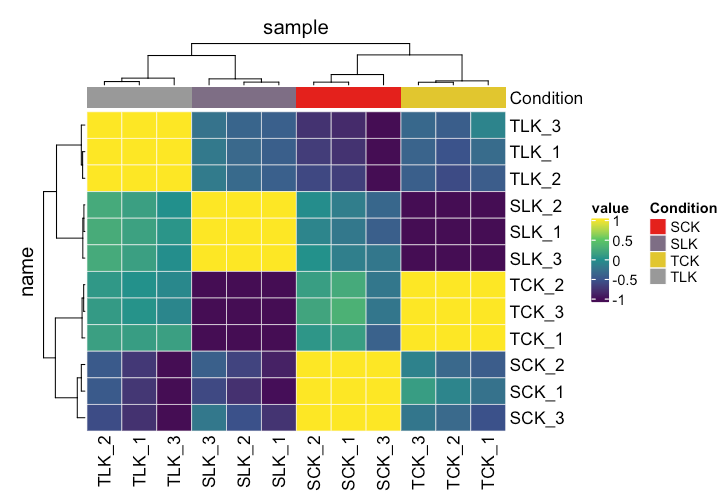
多些选择不是坏事
count数据归一化
数据归一化需要首先把矩阵转化为长透视表(gather或者pivot_long),然后按照样本名→转录本ID→表达量传入数据(tidybulk(sample, transcript_id, counts))。感觉再凑一个可以叫
RNA-seq连五鞭
泰迪大礼包
根据版本的不同(1.2+或者1.0+版本)使用不同方式标记表达丰度(低表达)和表达量归一化(normalization)。
counts_norm <- count_matrix %>%
#长透视表
gather(sample, counts, -transcript_id) %>%
#数据转化为tibble型
as.tibble() %>%
#数据装入tidybulk。样本名→转录本ID→表达量
tidybulk(sample, transcript_id, counts) %>%
#增加分组列condition
mutate(condition = sub("\\_\\w", "\\1", sample))
if (packageVersion("tidybulk") >= 1.2) {
counts_norm <-
counts_norm %>%
#按照condition分组筛选低表达基因
identify_abundant(factor_of_interest = condition) %>%
scale_abundance()
} else {
counts_norm <-
counts_norm %>%
scale_abundance(factor_of_interest = condition)
}
counts_norm %>%
ggplot(aes(counts_scaled + 1, group = sample, color= condition)) +
geom_density() +
scale_x_log10() +
theme_minimal()
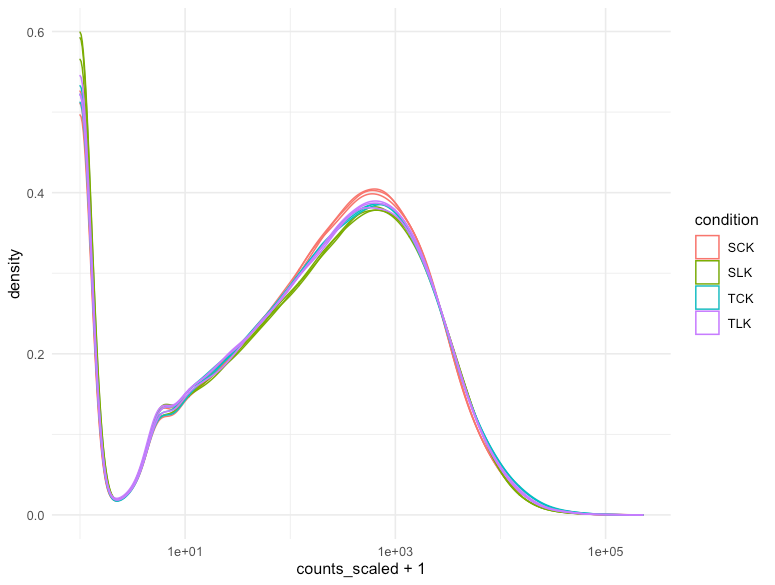
本次使用的数据集在这方面没有表现出很大的差异性。做不做归一化差别不大……
表达矩阵热图
接下来画出表达量差异最大的五百基因做一个热图来看看处理和品种间差异,tidybulk同样对流程进行了优化。
counts_norm %>%
#去除低丰度基因
filter(.abundant) %>%
#表达量差异最大的五百基因
keep_variable(.abundance = counts_scaled, top = 500) %>%
heatmap(
.column = sample,
.row = transcript_id,
.value = counts_scaled,
transform = log1p,
show_row_names = FALSE
) %>%
add_tile(condition)
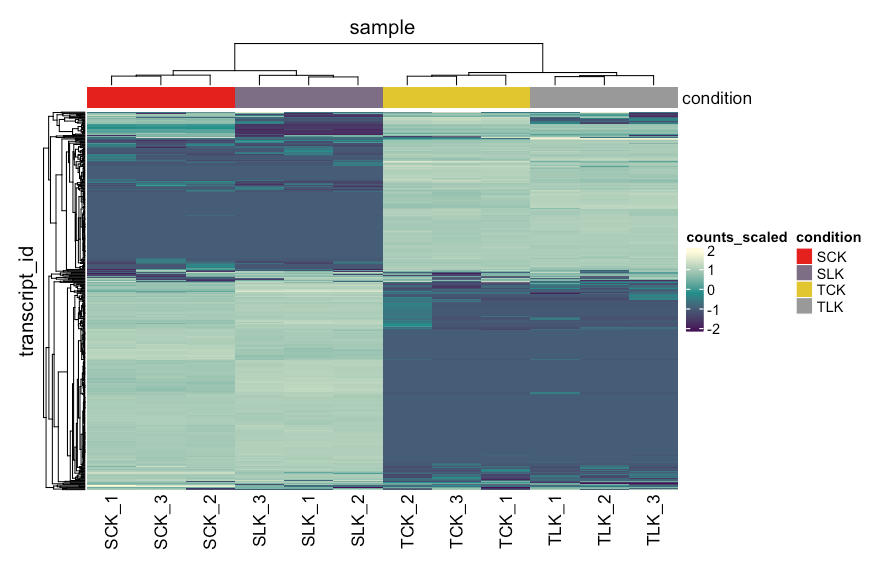
聚类
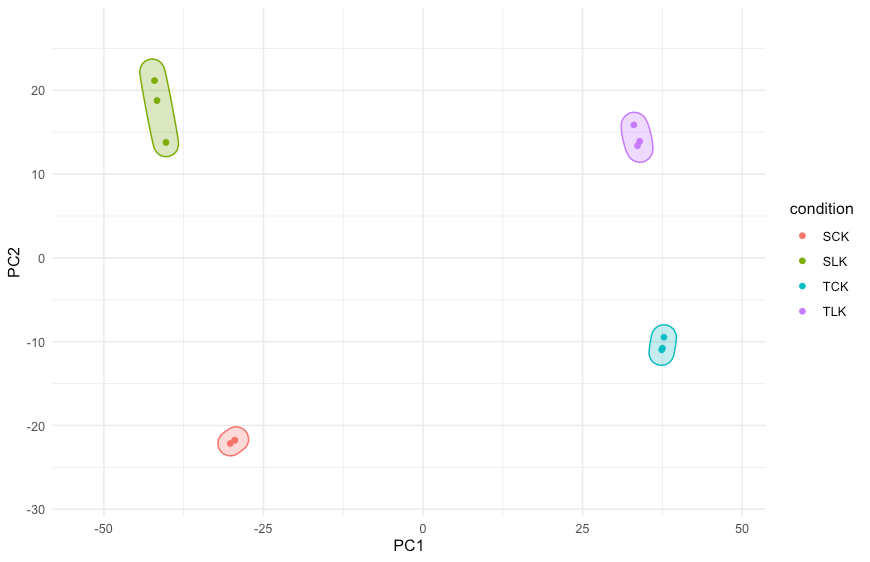
对于数据聚类,tidybulk提供两种方法: PCA聚类
tt.norm.PCA <-
counts_norm %>%
#PCA法,10个纬度,表达差异最大的2000个基因
reduce_dimensions(method="PCA", .dims = 10, top = 2000)
tt.norm.PCA %>%
pivot_sample() %>%
select(contains("PC"), everything())
tt.norm.PCA %>%
pivot_sample() %>%
select(starts_with("PC"), condition) %>%
GGally::ggpairs(columns = 1:10, ggplot2::aes(colour= condition))
这个图意义嘛……徒增功耗!最大的贡献肯定是来自PC1和PC2,剩下的似乎没必要做出来。

tt.norm.PCA %>%
pivot_sample() %>%
ggplot(aes(x = PC1, y = PC2, color = condition)) +
geom_point() +
theme_minimal() +
ggforce::geom_mark_ellipse(
aes(fill = condition),
show.legend = FALSE,
expand = unit(3, "mm")
) +
scale_x_continuous(expand = expansion(c(.2,.2))) +
scale_y_continuous(expand = expansion(c(.2,.2)))
MDS聚类
tt.norm.MDS =
counts_norm %>%
reduce_dimensions(method="MDS", .dims = 10)
tt.norm.MDS %>%
pivot_sample() %>%
select(contains("Dim"), condition) %>%
GGally::ggpairs(columns = 1:10, ggplot2::aes(colour=`condition`))
tt.norm.MDS %>%
pivot_sample() %>%
ggplot(aes(x = Dim1, y = Dim2, color = condition)) +
geom_point() +
theme_minimal() +
ggforce::geom_mark_ellipse(
aes(fill = condition),
show.legend = FALSE,
expand = unit(3, "mm"))+
scale_x_continuous(expand = expansion(c(.2,.2))) +
scale_y_continuous(expand = expansion(c(.2,.2)))
因为结果差不多,这里就不展示了
基因差异表达
test_differential_abundance集成了edgeR,limma和DESeq2三种方法,这里主要介绍edgeR和DESeq2。看作者源代码,DESeq2支持的一般。
counts_edge <- counts_norm %>%
test_differential_abundance(
.formula = ~ 0 + condition,
.contrasts = list(
c("conditionSLK - conditionSCK"),
c("conditionTLK - conditionTCK"),
c("conditionTLK - conditionSLK")
))
# counts_deseq <- counts_norm %>%
# test_differential_abundance(
# method = "DESeq2",










