三代数据解读
三代测序又被称为单分子实时DNA测序,采取纳米孔的单分子读取技术,相较之前测序技术,其优势为:读长长(20 Kb文库通常平均长度能达到8 Kb以上,如果DNA提取质量很高平均长度可以达到10 Kb以上),最长读长可以达到40 Kb~70 Kb左右;无需进行PCR扩增,避免reads复制比例(二代文库通常会出现复制比例偏高,覆盖度不均匀的情况);避免GC偏好性,高GC区域可以轻松测出来,对于那些高GC或低GC物种保证从头组装基因组的可靠性。
好了废话不多说,进入今天主题.
一.数据下机结果评估
1) RSII数据下机后会有一个质控报告,小编作为一个分析人员,比较关注的点有1、SMRT Cells测了几个cell;2、Mean Read Length的平均长度,长度过短可以说明这个文库构建失败了;3、Post-Filter后的数据量,这里Pre-Filter是没有进行任何处理的原始下机数据统计,Post-Filter是对原始数据进行过滤低质量和长度小于500 bp后的数据统计(官方标准);最后一个就是reads的分布情况。示意图如下:
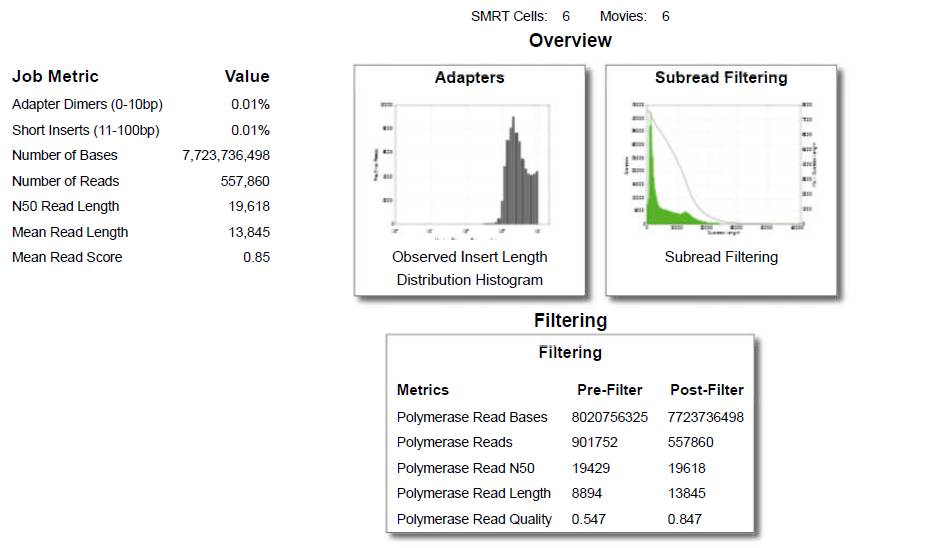
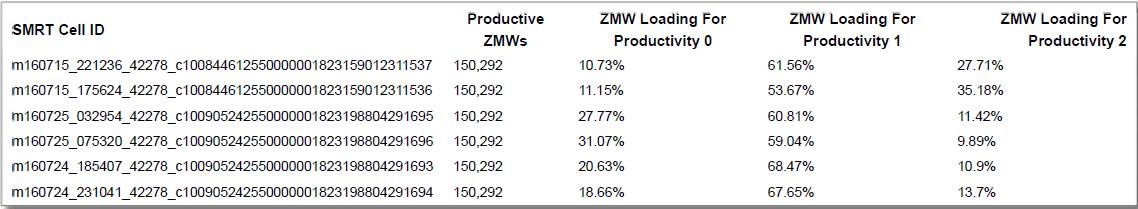
还需要注意的就是下面这张图,ZMW Loading For Productivity 1里面测的数据才是可用,但是理论上来讲P0~P2的比例为1:1:1(P0代表ZMW小孔中没有DNA掉落进去,P1只有一条进入,P2就是有多条以上),但是为了保证数据产出P1比例会高一些。
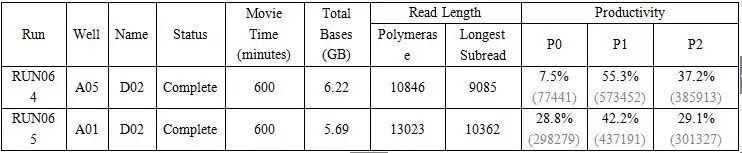
2)PacBio的新机器Sequel的数据下机暂时没有报告生成,需要拿到Subreads数据后自行进行处理统计。下面的这个表作为最初的一个参考,Total Bases产出数据量,Polymerase酶读长,以及P1的比例。
二. 数据形式
1)RSII数据是以*.h5,文件的格式进行存储,它是一个二进制的文件,通常情况是不太容易看的,所以通过软件对其进行处理成我们通常使用的FASTQ和FASTA格式。每一个cell都有对应的名字,点进去看到一些文件(信息统计结果)和一个Analysis_Results的文件夹(数据的藏身之所),这里小编借用别的小伙伴的图片更加直观展示。其中的subreads.fast*的数据是已经处理过了,可以将这三个直接合并到一起,用于分析。

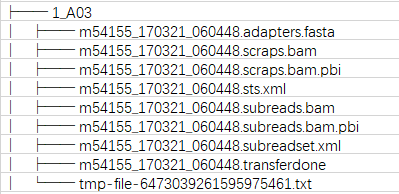
2)说完RSII那么Sequel的数据是一个怎样形式呢,这里Sequel平台一个cell的数据产出大约是RSII的4~5倍,面对这么大的数据量没有继续使用h5文件进行存储,直接采用的bam文件,它的文件目录与RSII区别还是非常大,不多说了直接上图。其中的adapters.fasta是接头序列,subreads.bam这个文件是已经去除测序接头后的数据,通过一些工具给它转化成fastq或fasta即可用于后面的分析,Xml文件是对其统计信息,transferdone是告诉人们我的这个cell成功测完了。
三.小结
Sequel产出的数据量在5Gb左右,是RSII的4倍左右,价格也相对便宜很多。不久将来PacBio公司会推出更加高通量的仪器,非常期待那个时候的下机数据会与RSII和Sequl有什么不同。
整理自百迈客公众号,有删改。











