
第一作者:田金树,孔儒,邓彬
通讯作者:李小年,朱艺涵,孙土来
通讯单位:浙江工业大学化学工程学院
论文DOI:10.1002/anie.202409556
烷烃直接脱氢是工业制烯烃的重要途径,Pt基催化剂是工业上最常用的催化体系,但却面临着催化剂失活的重要挑战。常见的失活机制往往包括金属颗粒的烧结、催化剂表面的积碳等。在排除这些机理以外,在PtZn等Pt基合金催化体系中,往往还存在着反应初期快速失活的现象。因此解析该过程的失活机制以及提出全新应对策略对于设计高稳定性的丙烷脱氢催化剂至关重要。
近日,浙江工业大学化工学院李小年、朱艺涵教授团队在该方面取得重要进展。以负载PtZn合金催化剂为研究对象,结合电子显微镜技术和原位谱学技术从微结构视角揭示了该类合金催化剂失活的新机制。该非经典失活机制来自于反应条件下Zn在合金中的自发迁移所诱导的PtZn到Pt
3
Zn相变。基于对相关失活微观机制的认知提升,研究团队提出了载体-金属界面化学微环境的调控策略,并通过削弱表面羟基和金属的相互作用力,成功抑制了合金相中Zn的迁移,最终在丙烷脱氢的过程中大幅缓解了催化剂由非经典失活机理带来的失效现象。
丙烯是一种重要的化工原料,但是传统的从石脑油中生产丙烯的工艺已经不能满足日益增长的丙烯需求。因此急需开发新的丙烯生产方式,而丙烷直接脱氢(PDH)技术作为一种高效直接的生产丙烯的技术受到了广泛的关注。PDH反应通常使用Pt 基催化剂,温度约为600℃。然而,这些催化剂在反应过程中经常由于焦炭积碳和活性金属烧结而失活。为了应对这些挑战,一种常见的方法是引入第二种金属,与活性金属 Pt 形成合金,以抑制催化剂的焦炭沉积和烧结。与纯 Pt 相比,Pt 与其他金属(如 Ga、In、Co、Cu 和 Zn)的组合表现出了更优异的脱氢性能。其中Zn 作为一种无毒且储量丰富的金属,由于其在 PDH 反应中与 Pt 配合使用时表现出色,因此作为双金属系统中的首选的第二种金属而引起了广泛关注。但是PtZn
IMA仍然面临着失活的问题,因为对催化剂结构演变的微观理解有限,所以解决失活问题变得尤其复杂。因此全面解析催化剂在运行过程中微观结构的改变能够有效启发对催化剂自下而上的设计策略,对构建更高效稳定的催化剂具有重要意义。
1. 结合电子显微镜技术和原位谱学技术从微结构视角揭示了该类合金催化剂失活的新机制。该非经典失活机制来自于反应条件下Zn在合金中的自发迁移所诱导的PtZn到Pt
3
Zn相变。
2.
提出了载体-金属界面化学微环境的调控策略,并通过削弱表面羟基和金属的相互作用力,成功抑制了合金相中Zn的迁移,最终在丙烷脱氢的过程中大幅缓解了催化剂由非经典失活机理带来的失效现象。
PDH反应通常使用铂基催化剂在600℃左右的温度下进行。然而,这些催化剂常因反应过程中积碳和活性金属的烧结而失活(图1A)。在这里,作者发现了在用于PDH的PtZn/γ-Al
2
O
3
催化剂中观察到的独特失活机制。对于Pt基 IMA PDH系统,催化剂存在与第二组份分离、迁移和重构的问题(图 1B)。这些变化直接影响催化活性位点,导致催化剂失活。通过一系列表征揭示了催化剂失活是由PtZn纳米相转变为 Pt
3
Zn纳米相并伴随脱锌作用引发的。γ-Al
2
O
3
载体上的羟基与Pt-Zn合金上的Zn位点之间的强相互作用促进了这种转变。最终,提出了一种创新策略,通过修改γ-Al
2
O
3
载体的表面来减轻催化剂失活(图 1C)。这种修饰削弱了金属与载体的结合,从而减少了 PtZn IMA 的脱锌并有效抑制了催化剂失活。
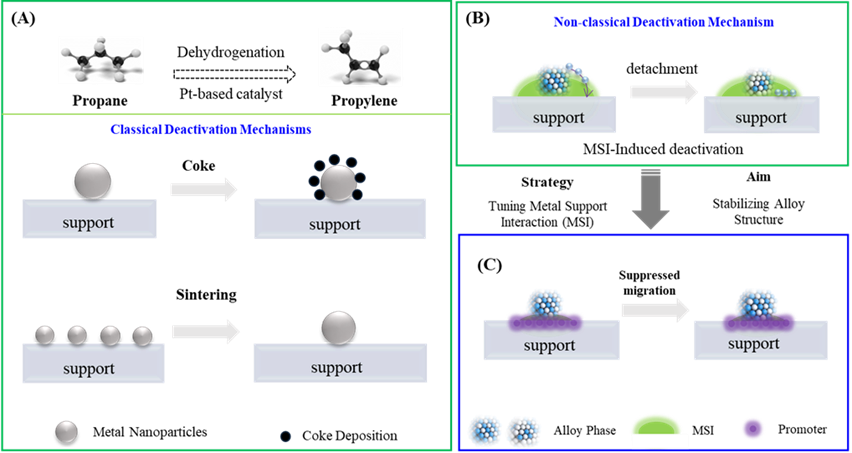
图1. A,Pt基催化剂PDH的经典失活机理。B,非经典金属-载体相互作用(MSI)诱导的表面重构失活机理。C,调控MSI实现合金结构稳定性的示意图。
图2展示了不同催化剂(即Pt/γ-Al
2
O
3
、PtZn/γ-Al
2
O
3
和PtZn/0.5K-γ-Al
2
O
3
)在PDH反应中的性能。最初,纯 Pt 基催化剂 Pt/γ-Al
2
O
3
在PDH反应中表现出较高活性(图 2A)。然而,该催化剂会经历连续失活,丙烯选择性较差,低于约 80%。另一方面,PtZn/γ-Al
2
O
3
在PDH反应中表现出较高的初始活性(图 2B),但在前30分钟内迅速失活。在剩余的测试时间内,活性稳定在20%左右的转化率。值得注意的是,该催化剂在整个测试过程中始终保持对丙烯的高选择性,约为 93%。这凸显了Zn在PDH反应中的关键作用,通常归因于PtZn/γ-Al
2
O
3
催化剂中PtZn IMA 的形成。令人惊讶的是,与PtZn/γ-Al
2
O
3
相比,PtZn/0.5K-γ-Al
2
O
3
催化剂表现出更显着的失活抑制作用(失活速率从0.2044 h
-1
降低到0.0587 h
-1
),特别是在操作的最初30分钟内。在 590℃的300分钟测试中,该催化剂保持了卓越的性能,丙烷转化率约为 34%,丙烯选择性约为 95%,WHSV为54 L/g
cat
·h(图 2C)。

图2. Pt/γ-
Al
2
O
3
(A)、PtZn/γ-
Al
2
O
3
(B) 和 PtZn/0.5K-γ-
Al
2
O
3
(C) 中丙烷转化率和丙烯选择性随反应时间的变化。
为了探究催化剂失活机制,利用HAADF-STEM对反应前后负载型IMA催化剂进行明确的原子结构研究,揭示了IMA的原子级堆积和组成。我们成功地在不同区域对新鲜和反应后的PtZn/γ-Al
2
O
3
催化剂进行成像(图 3)。从图 3(A-B) 可以清楚地看出,新鲜的PtZn IMA沿[112]和 [100]方向的STEM图像表现出有序的原子堆积和交替的行,归因于Pt和Zn形成IMA,与其各自的结构投影和采用P4/mmm空间群的四方PtZn IMA相的模拟STEM图像匹配良好(图 3A-B)。这些观察结果明确证实了合成的PtZn/γ-Al
2
O
3
催化剂中存在四方PtZn IMA。此外,使用能量色散X射线光谱 (EDS) 的元素图谱证实了Pt和Zn在整个PtZn IMA纳米粒子中的均匀分布,如图3E所示。相比之下,HAADF-STEM 在废PtZn/γ-Al
2
O
3
催化剂上显示出另一种Pt-Zn合金结构(图3C-D和图S15G-L)。在图3C-D中,STEM 图像表现出由Zn决定的对比度,其对比度较暗,周围环绕着具有明亮对比度的四角形排列或六角形排列的Pt,这与[100]和[111]结构投影以及模拟的STEM图像非常相似。为立方Pt
3
Zn IMA相,空间群为Pm
̅
3 m(图 3C-D)。通过细致的原子级实验观察,我们提供了令人信服的证据,证明PDH过程中PtZn/γ-Al
2
O
3
催化剂中从四方PtZn到立方Pt3Zn IMA相的不可逆相变。这种相变与脱锌过程密切相关,可能对 PtZn/γ-Al
2
O
3
催化剂的失活产生关键影响。此外,我们发现Zn元素位于载体表面,并且分散在Pt
3
Zn IMA相内部(图 3F),这与新鲜PtZn/γ-Al
2
O
3
催化剂中观察到的情况显着不同。这意味着 PtZn IMA 表面上的 Zn 元素可能会发生迁移,导致其在反应过程中向载体表面移动。有趣的是,我们发现无论是新鲜的还是用过的催化剂,K 改性的Al
2
O
3
样品上的Zn元素分布主要集中在Pt原子附近。载体上没有检测到 Zn 元素的存在,而载体表面主要为K元素(图 3G,H)
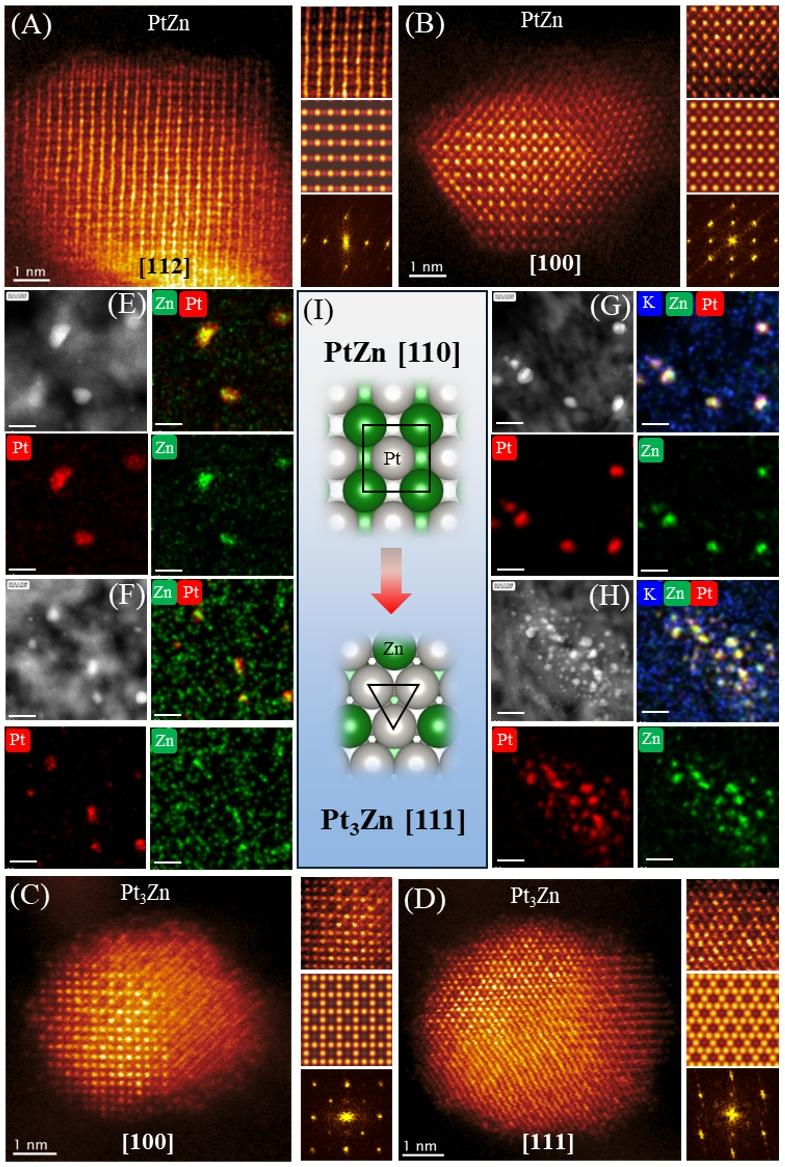
图3.
(A-B) 新鲜PtZn/γ-Al
2
O
3
和 (C-D) 废PtZn/γ-Al
2
O
3
原子分辨率HAADF-STEM图像。(E) 新鲜PtZn/γ-Al
2
O
3
、(F) 废PtZn/γ-Al
2
O
3
、(G) 新鲜PtZn/0.5K-γ-Al
2
O
3
和 (H) 废 PtZn/0.5K-γ-Al
2
O
3
(Pt,红色;Zn,绿色;K,蓝色) 的低倍HAADF-STEM图像和相应的EDS元素图。(I) 使用原子结构模型描绘从 PtZn 到 Pt
3
Zn IMA 相的相变示意图。
为了进一步了解反应过程中PtZn合金的纳米相变,我们对不同反应时间的PtZn/γ-Al
2
O
3
和PtZn/0.5K-γ-Al
2
O
3
样品进行了CO-DRIFTS实验(图 4)。在2000 ~ 2100 cm
-1
振动频率范围内观察到的峰值表明CO在Pt位点上的线性吸附。在~1860 和 1950 cm
-1
之间发现的峰归因于CO在多Pt原子上的桥吸附峰。有趣的是,在整个反应过程中,PtZn/γ-Al
2
O
3
催化剂上没有检测到CO桥式吸附峰。这表明Zn的添加促进了Pt原子的分散,防止了Pt簇或纳米颗粒的形成。图 4A说明了PtZn合金上CO特征吸附峰的明显变化。具体来说,在1分钟和3分钟内观察到的 PtZn/γ-Al
2
O
3
催化剂的主振动峰位于2045 cm
-1
,这归因于PtZn IMA上吸附的顶部CO。然而,随着反应时间进行到5分钟及更长,主振动峰转移到2071 cm
-1
。CO主振动峰的这种变化表明CO与Pt-Zn合金纳米相之间的结合强度发生变化。前期研究表明,合金化的Zn原子可以增加Pt位点的电子密度,导致红外频率向低频移动。这表明Pt-Zn纳米相中Zn的减少导致Pt电子密度降低。与PtZn IMA相比,Pt
3
Zn IMA周围的Zn数量较少,导致Pt的配位和电子环境发生变化。对于PtZn/K-γ-Al
2
O
3
系统,我们观察到不同的现象,如图4B所示。我们发现,归因于PtZn IMA的Pt
1
位点(~2049 cm1)对应的峰位置在整个反应期间(1 - 30 分钟)保持相对稳定。
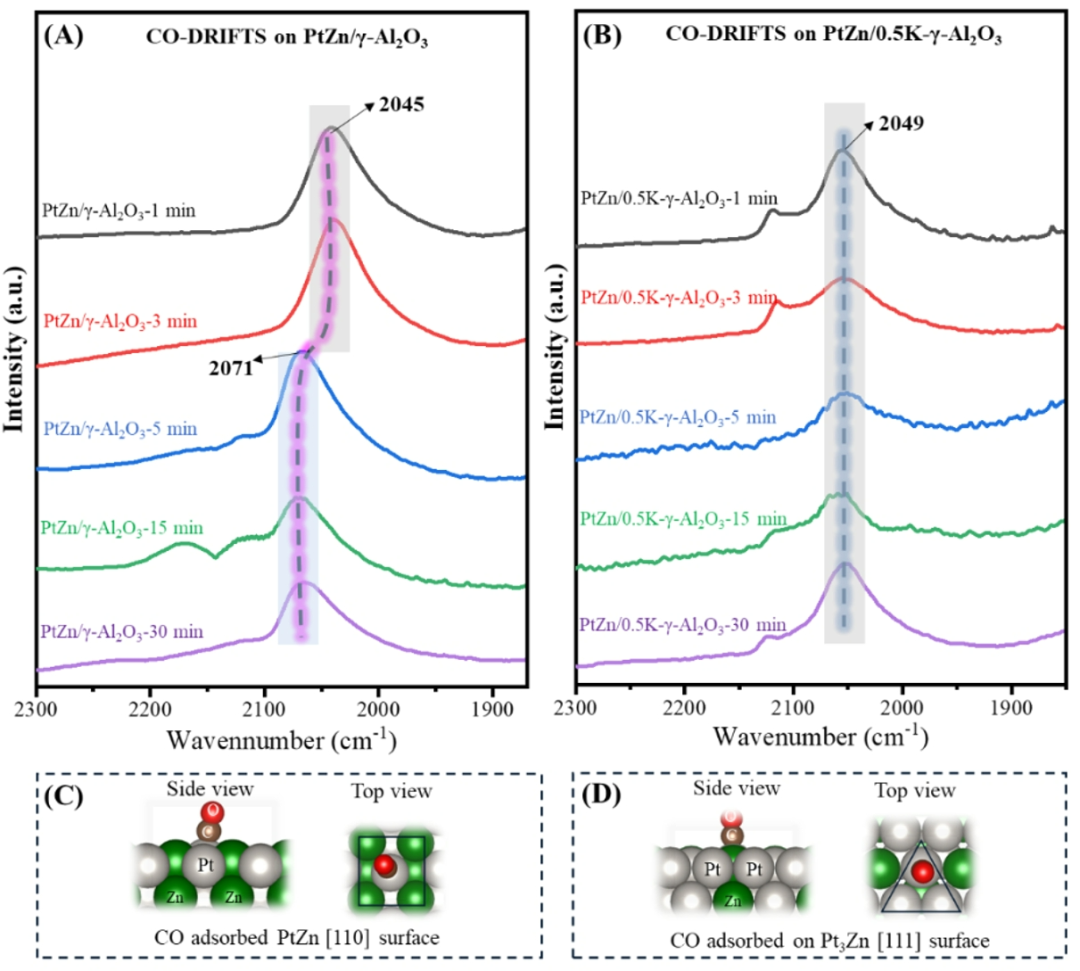
图4.
(A-B)不同反应时间的PtZn/γ-Al
2
O
3
和PtZn/0.5K-γ-Al
2
O
3
催化剂上的CO漫反射红外傅里叶变换光谱 (CO-DRIFTS)。(C-D) CO 吸附在 Pt
1
Zn 和 Pt
3
Zn IMA 上的示意图。
为了进一步阐明反应过程中Zn脱离的驱动力,进行了原位傅里叶红外实验(原位 FTIR)并在图5中进行了讨论。1500 cm
-1
处的峰代表吸附的C
x
H
y
物质的C-H键的振动,3400-3700 cm
-1
之间的峰归因于γ-Al
2
O
3
表面羟基的振动。随着反应的进行,我们观察到表面C-H键振动的强度逐渐降低,表明催化性能下降。这一观察结果与图2中所示的实验结果一致。此外,我们注意到载体表面上的羟基呈下降趋势,表明这些物质在PDH反应过程中逐渐被消耗。我们认为羟基的减少是由于Pt-Zn合金中的Zn原子结合的羟基驱动的。因此,我们提出了在γ-Al
2
O
3
表面上Pt-Zn合金重构的机制,如图5B所示。最初,部分Zn物质在反应条件下从PtZn IMA表面分离。随后,这些剥离的Zn物质向γ-Al
2
O
3
载体迁移,众所周知,γ-Al
2
O
3
表面上的这些羟基对捕获Zn物种具有很强的亲和力,从而导致ZnO
x
H
y
物种的形成。Zn分离和表面ZnO
x
H
y
物质形成之间存在一定的平衡。最终,Zn含量较低Pt
3
Zn IMA形成并在γ-Al
2
O
3
表面保持稳定。我们还进行了计算,测量Zn原子在具有羟基的γ-Al
2
O
3
表面上的Pt-Zn合金内移动所需的能量(图 5C)。利用稳定且大量暴露的γ-Al
2
O
3
(110)晶面进行建模,模拟Zn的迁移以及K在抑制Zn迁移中的作用。当从PtZn合金中分离出来时,Zn原子很容易与羟基结合生成ZnO
x
H
y
物质。如图所示,ZnO
x
H
y
形成的能量趋势为-2.54
eV,表明这种转变非常容易发生。对PtZn/K-γ-Al
2
O
3
进行了原位FTIR,结果显示,在整个反应过程中,在 3250 cm
-1
和3750 cm
-1
之间观察到的峰保持相对稳定(图 5D),这表明羟基是反应过程中不被消耗。从上述讨论中可以看出,催化失活与 PtZn IMA 的重构密切相关,而 PtZn IMA 的重构又受到γ-Al
2
O
3
表面羟基的影响。因此,可以通过改变γ-Al
2
O
3
表面的羟基来调节活性位点,从而提高催化剂的稳定性。羟基通过氢键相互作用,导致主要源自这些氢键羟基的弱路易斯酸位点的存在。因此,有人提出利用钾(K)等碱性物质来调节γ-Al
2
O
3
的表面微环境并调节PDH的性能。适当的K改性可以调节金属与载体之间的相互作用(MSI),有效抑制Zn物种的迁移并保持粒径,有效平衡了PtZn纳米颗粒烧结和表面Zn迁移的影响,从而实现最佳的催化活性。基于这一现象,我们提出了一个示意图(图5E),强调通过使用K调节γ-Al
2
O
3
微环境来稳定PtZn IMA的概念。通过占据γ-Al
2
O
3
表面上的羟基,K离子的存在有效防止Zn的脱离,从而实现PtZn IMA所需的稳定性。进行了理论计算,以确定锌在K改性Al
2
O
3
表面迁移形成 ZnO
x
H
y
物质所需的能量(图 5F)。与K-γ-Al
2
O
3
(110)表面 (0.98 eV)相比,锌在 PtZn/γ-Al
2
O
3
(110)表面 (-2.54
eV)上更容易迁移。
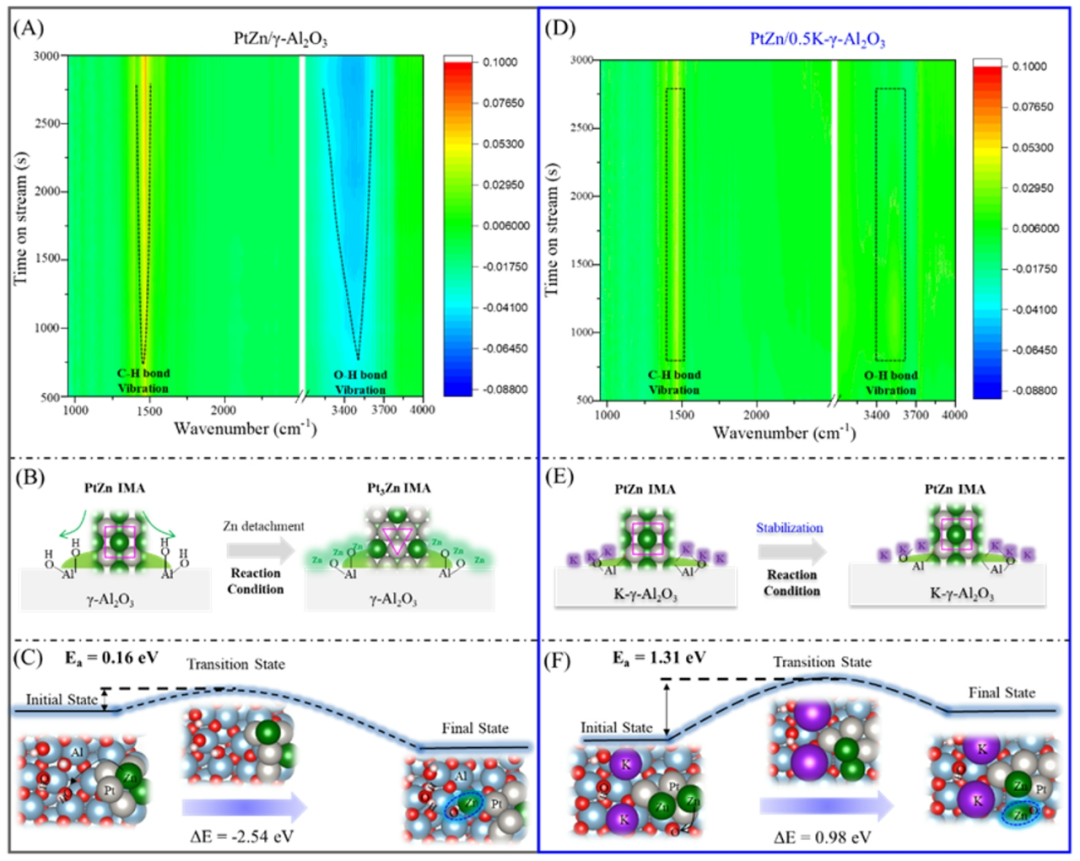
图5. (A和D) PtZn/γ-Al
2
O
3
和PtZn/0.5K-γ-Al
2
O
3
催化剂在PDH反应过程中的原位红外光谱;(B和E)
γ-Al
2
O
3
和0.5K-γ-Al
2
O
3
表面Zn从PtZn到Pt
3
Zn
IMA的分离和稳定过程示意图;(C和F) Zn从Pt-Zn结构分离到载体上的能量变化,B为PtZn/γ-Al
2
O
3
,E为PtZn/0.5K-γ-Al
2
O
3
。
本工作以负载PtZn合金催化剂为研究对象,结合电子显微镜技术和原位谱学技术从微结构视角揭示了该类合金催化剂失活的新机制。该非经典失活机制来自于反应条件下Zn在合金中的自发迁移所诱导的PtZn到Pt
3
Zn相变。基于对相关失活微观机制的认知提升,研究团队提出了载体-金属界面化学微环境的调控策略,并通过削弱表面羟基和金属的相互作用力,成功抑制了合金相中Zn的迁移,最终在丙烷脱氢的过程中大幅缓解了催化剂由非经典失活机理带来的失效现象。
李小年
,加拿大工程院院士、教育部长江学者特聘教授。主要从事工业催化剂技术开发与绿色工艺创新研究,主持国家重点研发计划重点专项、国家自然科学基金、浙江省自然科学基金等省部级及以上项目20余项,先后获国家技术发明二等奖2项(分别排名第1、第3),国家科学技术进步一等奖1项(排名第5)、省部级科技成果一等奖4项和中国青年科技奖等,发表学术论文500余篇,获授权发明专利100余项。
朱艺涵
,国家级青年人才/教授,化工学院副院长。主要从事先进电子显微方法学发展和物质科学应用,相关成果在Science及其子刊、Nature子刊等权威期刊发表论文140余篇,引用次数17000余次,H指数64。先后主持国家优秀青年科学基金、面上基金等4项,省杰出青年科学基金1项,承担国家重点研发计划重点专项课题1项。
田金树
,运河青年学者,特聘教授,主要从事低碳分子高值化利用,结合从头算分子动力学、原位动态表征技术以及微观动力学,识别催化反应过程活性中心和反应机理。主持国自然基金-面上项目1项,省属高校基本科研业务费(科技类)项目1项;相关成果部分发表在Sci. Adv.、Nat. Commun.、Angew. Chem. Int. Edit 、J. Am. Chem. Soc.,ACS Catalysis 等国际权威期刊。







