鬼臼毒素的结构修饰及抗肿瘤活性研究进展
Research Progress of the Structural Modification of
Podophyllotoxin and Its Antitumor Activities
成伟华1,2,陈 虹3,邹忠梅1*
(1. 中国医学科学院北京协和医学院药用植物研究所,北京 100193;2. 原子高科股份有限公司,北京 102413;
3. 武警后勤学院生药学教研室,天津 300162)
摘要:
芳基萘类木脂素鬼臼毒素是从不同鬼臼属植物根茎中提取的树脂中的主要成分,具有显著的抗肿瘤活性。但是
由于毒性大限制了其作为抗肿瘤药物的应用,因此对鬼臼毒素开展了大量结构修饰工作。依托泊苷(VP-16)和替尼泊苷(VM-26)作为鬼臼毒素衍生物,现已广泛用作抗肿瘤的临床治疗。然而,临床应用发现依托泊苷和替尼泊苷水溶性差,易产生多药耐药和严重的胃肠道功能紊乱,为了克服上述缺陷,寻找更有效的鬼臼毒素类衍生物显得尤为迫切。本文对鬼臼毒素抗肿瘤作用机制、结构修饰和构效关系进行了总结,为新鬼臼毒素衍生物的合成提供参考。
关键词:
鬼臼毒素衍生物;作用机制;结构修饰;构效关系;抗肿瘤活性;合成工艺
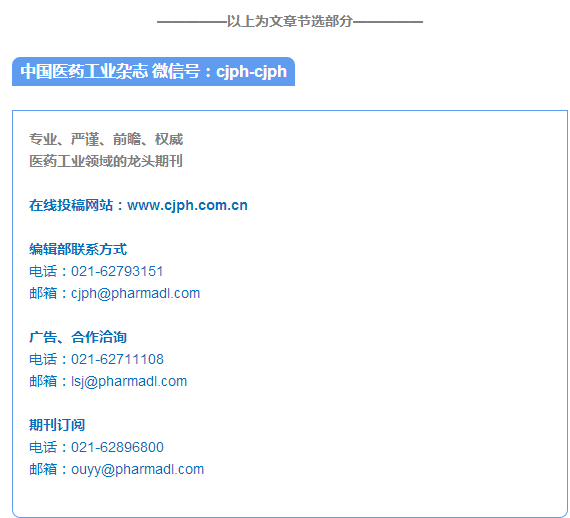
鬼臼毒素(podophyllotoxin,1) 又称鬼臼素、
鬼臼毒、鬼臼脂素,是从鬼臼属植物Dysosma 中分离得到的一种芳基萘类木脂素内酯,具有抗肿瘤、抗病毒、抗炎等多种生物活性,其中抗肿瘤活性最为显著。但是1 对胃肠道有严重不良反应,不适于直接在临床上使用。从上世纪50 年代开始,研究人员对1 进行了大量结构修饰工作,其衍生物依托泊苷(etoposide,VP-16,2) 和替尼泊苷(teniposide,VM-26,3) 已成功上市,成为临床抗肿瘤一线治疗药物,但是仍然存在严重的不良反应,如易产生胃肠紊乱和骨髓抑制等。经过不懈努力,陆续发现了一些具有更佳药理学和药代动力学性质的新型1 衍生物,如NPF(4)、GL331(5) 和NK611(6) 等( 图1)。近年来,随着分子肿瘤学、分子药理学对肿瘤
本质的逐步阐明,人们在1 及其衍生物的抗肿瘤机
制、结构修饰及构效关系(SAR) 研究等方面取得了很大的进展。本文将从1 的抗肿瘤作用机制、各环的结构修饰和构效关系3 个方面进行综述。
1 1 衍生物抗肿瘤机制
1 衍生物具有高抗肿瘤活性,已受到越来越多
的关注。对该类化合物抗肿瘤机制的阐明有利于对其进一步开展结构优化和发现新型先导化合物。现有研究表明,1 及其衍生物具有多种抗肿瘤作用机制,主要有抑制微管蛋白聚合、抑制拓扑异构酶Ⅱ活性、自由基机制等。
1.1 抑制微管蛋白聚合
1 及其衍生物可以与微管蛋白结合,抑制微管
聚合,进而导致染色体在复制以后不能分离,细胞分裂停止在G2/M 期,使肿瘤细胞的分裂难以正常进行,继而发挥抗肿瘤的作用[1]。
1.2 抑制拓扑异构酶Ⅱ活性
抑制拓扑异构酶Ⅱ活性可以阻止肿瘤细胞快
速生长增殖,进而杀死肿瘤细胞。现今,拓扑异构酶Ⅱ已成为多种抗肿瘤药物的作用靶点[2]。研究发现2 是作用于细胞核中的拓扑异构酶Ⅱ从而引起DNA 的变化,继而发挥抗肿瘤作用。2 主要以共价键的形式与DNA 和拓扑异构酶Ⅱ形成稳定的“药物-DNA- 拓扑异构酶Ⅱ”三元复合物,使DNA 在复制过程中发生异常重组,抑制DNA 连接活性,使
DNA 双链发生断裂,最终导致肿瘤细胞周期停滞在
G0/G1 期,使其生长繁殖受到抑制,并引起凋亡相关蛋白酶的激活和表达[3]。进一步研究发现,2 的抗
肿瘤作用主要与抑制拓扑异构酶Ⅱ α 型有关[4]。
1.3 自由基机制
自由基的形成可能是诱导DNA 断裂的原因,
这种感应能被自由基捕获剂所抑制[ 5],但是这种活性物质在拓扑异构酶Ⅱ介导的DNA 断裂中的作用,以及自由基-DNA 加合物机制与拓扑异构酶Ⅱ毒性机制之间的关系还不是很清楚。此外,van Maanen等提出4′- 去甲基表鬼臼毒素衍生物的细胞毒作用有可能是E 环在体内代谢过程中形成邻苯二酚、邻醌式结构和苯氧自由基形式,这些活性物质可以和DNA 形成稳定复合物,从而使DNA 损伤。因此认为自由基代谢物可能对4′- 去甲基表鬼臼毒素衍生物抗肿瘤活性起一定作用[6]。
2 1 的化学修饰
2.1 A 环结构修饰
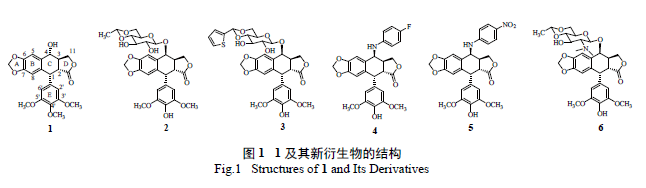
上世纪60 年代到90 年代,对1 的A 环进行了
一系列结构修饰,结果发现活性都比母体化合物有所下降。2003 年Castro 等对1 的A 环进行结构修饰[7],合成了异噁唑鬼臼毒素类衍生物7( 图2),同样,这些衍生物的活性均弱于母体化合物。因此近几年对A 环的结构修饰报道越来越少,说明对A环的修饰已经不再是一个热点。
2.2 B 环结构修饰
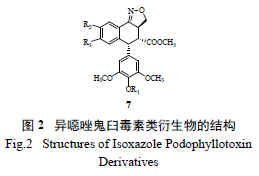
对B 环直接修饰的报道很少,都是基于天然
产物α- 盾叶鬼臼素和β- 盾叶鬼臼素来进行的。Thurston 等合成了C-5 位氧取代系列衍生物8 或糖苷化衍生物9( 图3) [8],活性测定显示所合成衍生物对KB 细胞有不同程度的细胞毒活性,其中衍生
物8 中一个化合物的活性是2 活性的100 倍,而
C-5 位连接糖基的衍生物9 却只显示微弱的细胞毒活性,且对拓扑异构酶Ⅱ显示出很弱的抑制作用。
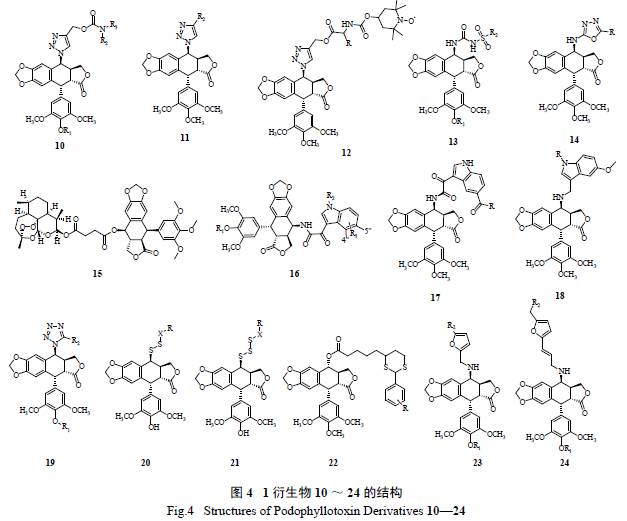
2.3 C 环的修饰( 图4)
对1 的C 环的结构修饰主要集中在C-4 位上,
包括C- 取代衍生物、O- 取代衍生物、S- 取代衍生物、Se- 取代衍生物和N- 取代衍生物,其中以N- 取代衍生物最多,结构也最为丰富,下面就近期代表性的C-4 位N- 取代和其他取代的1 衍生物进行介绍。三唑类化合物具有多种生物活性,包括抗真菌、抗病毒、抗过敏和抗肿瘤活性,已经成为研究的热点。当C-4 位用三唑取代时,既提高了活性,又增强了水溶性。Liu 等在C-4 位引入氨基甲酸酯取代的三唑后,得到一系列衍生物10[9],通过对HL-60、A-549、HeLa 和HCT-8 进行细胞毒性测试,结果表明合成的化合物活性均高于2,且体内代谢良好。另外Chen 等合成了一系列三唑衍生物11,大部分化合物具有显著的抗肿瘤活性,且具有较好的抗多药耐药活性[ 10]。Yang 等合成了自旋标记的三唑衍生物12,当取代基R 为异丙基时显示很好的细胞毒活性,并且对耐药细胞也有良好活性[11]。
Zhang 等合成了3 个系列的4β- 磺脲鬼臼毒素衍生物13,对A-549、DU-145、KB 和KBvin 这几种细胞株有显著的细胞毒性,并且对耐药的KB 细胞株也有良好的细胞毒性[ 12]。结果说明,磺脲的引入可能是提高抗多药耐药活性的原因之一。Ren等在C-4 位引入1,3,4- 噁二唑杂环,合成得到一系列化合物14,细胞毒性均高于2( IC50 值1.51 ~5.49 μmol/L)[13]。Zhang 等把青蒿酯和1 拼接在一
起得到15,结果发现其对多种肿瘤细胞具有较好的
细胞毒活性,并且可能由于青蒿素的引入,使其对耐药的肿瘤细胞也具有良好活性[14]。
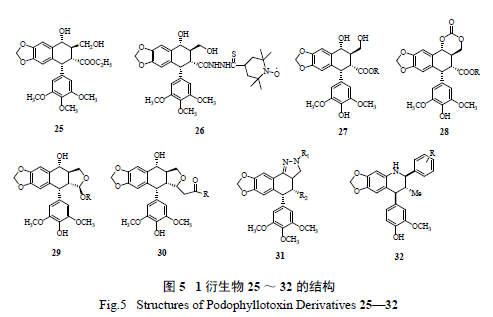
2.4 D 环的修饰( 图5)
大多数具有抗肿瘤活性的1 衍生物,具有一个
高张力的反式γ- 内酯环。该内酯环在热力学上是不稳定的,容易发生2- 位异构化生成苦鬼臼毒素。在体内2 的D 环2- 位异构化或内酯环开环后,抗肿瘤活性会显著降低,甚至没有活性[24]。
Gensler 等合成了一系列去内酯羰基,用碳环、
环氧醚、环酮及环硫醚取代内酯环的化合物[ 25],其细胞毒活性及其对细胞微管聚合作用均弱于母体化合物1。该课题组还合成了一系列2- 位氯代、氟代、羟基、甲基及溴取代的1 衍生物来考察内酯环的作用[26],发现只有2- 氯取代化合物在体内对P-388细胞显示明显的抗肿瘤活性。
2.5 E 环的修饰( 图6)
1 的E 环修饰分为:① E 环不同位置去氧化;
② 4'- 位去甲基化生成4'-OH,再通过不同方法得到各种4'- 位酯化衍生物。Hu 等合成了2'- 氯代鬼臼毒素衍生物33[ 33],其细胞毒活性均消失,对DNA 拓扑异构酶Ⅱ也没有抑制作用,且对细胞内微管蛋白聚合也没有破坏性。失去活性的原因可能是由于2'- 位氯代后,阻
止了E 环的自由旋转和E 环邻醌式结构的形成。虽
然在E 环连接不同的卤素抗肿瘤活性消失,但是其杀虫作用却有明显提高[34—36]。
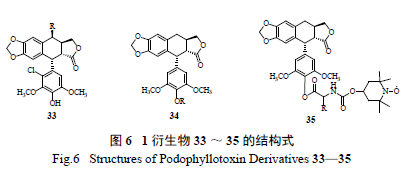
1 的4'- 位去甲基化生成4'-OH,然后可通过
不同方法得到各种4'- 位酯化衍生物。文献用脂肪酸、不饱和脂肪酸和氨基酸制备得到一系列4'- 去甲去氧鬼臼毒素衍生物34[ 37—39],尽管体外活性较母体化合物差,但在体内显示出明显的抗肿瘤活性。Zhang 等合成了氮氧自由基的4'- 去甲去氧鬼臼毒素衍生物35[40],与4'- 去甲基表鬼臼毒素和2 相比,这些化合物对HL-60、RPMI-8226 和A-549 细胞株具有更强的细胞毒活性。
3 总结与展望
综上所述,1 的结构修饰可在其所有环系进行。
大多数具有抗肿瘤作用的1 衍生物的A环是闭合的,B 环保留原有结构,C 环C-4 位连接不同取代基,D 环具有一个高张力反式γ- 内酯环,E 环中C-4' 位为羟基。D 环内酯环虽然对1 的生物活性起到很重要的作用,但是近年来研究发现,它并不是必须的,一些打开内酯环的1 衍生物仍然显示出很强的抗肿瘤活性。E 环中的C-4' 位羟基若被烷基、酰基所保护,则活性显著下降。只有当被封闭的C-4' 酚羟基被恢复,在体内与DNA 拓扑异构酶Ⅱ稳定结合,才有可能显示强抗肿瘤活性。尽管具有更佳药理学和药代动力学性质的新型1 衍生物相继被发现,如NPF、GL331、NK611、TOP53、Azatoxin和Etopophos 作为抗肿瘤新药已进入临床研究,且
这些化合物都表现出比依托伯苷更优越的活性,但
仍存在一些问题。相信随着对作用机制的深入研究和构效关系的更加完善,对1 衍生物的研究将进入更为深层次的领域。
作者简介:成伟华(1985—),男,助理研究员,从事天然产物结构
修饰及
放射性药物研究。
Tel:010-69358944
E-mail:[email protected]
通信联系人:邹忠梅(1964—),女,研究员,从事天然产物化学
研究。
Tel:010-57833290
E-mail:[email protected]