题记:这篇读书笔记介绍机器学习中分类问题最常用的一种解决办法:Logistic回归模型与判别分析。本文主要参考了Cory Lesmeister博士主编的《Mastering Machine Learning with R》(Second Edition)一书,这是一本机器学习的入门级教材。数据来自R语言MASS包自带数据集biopsy,代码来自
《Mastering Machine Learning with R》这本书配套的代码,笔者对部分代码进行修改。本文是个人的读书笔记,仅用于学习交流使用,我只是一个知识的搬运工。
1. 背景介绍
分类问题是机器学习的一大类问题,最好能使用0和1之间的概率进行建模,当然建模的方法很多。本文重点讨论Logistic回归模型与判别分析。Logistic回归就是以对数发生概率比为响应变量进行线性拟合,即log(P(Y)/1-P(Y)) = B0+B1。这里的系数是通过极大似然估计得到的。极大似然的直观意义就是,我们要找到一对B0和B1的估计值,使它们产生的对观测的预测概率尽可能接近Y的实际观测结果,这就是所谓的似然性。与其他软件一样,R语言也可以实现极大似然估计,通过找到最优的B值组合来使似然最大。
2. 数据准备
威斯康星大学的威廉·沃尔伯格博士1990年发布了一个来自威斯康星州乳腺癌患者数据集。目的是想明确肿瘤活检结果是良性还是恶性。研究者使用细针穿刺(FNA)技术收集样本并进行进行活检。
数据集包含699个患者的组织样本,保存在有11个变量的数据框中,如下所示:
ID:样本编码
V1:细胞浓度
V2:细胞大小均匀度
V3:细胞形状均匀度
V4:边缘黏着度
V5:单上皮细胞大小
V6:裸细胞核(16个观测值缺失)
V7:平和染色质
V8:正常核仁
V9:有丝分裂状态
class:肿瘤诊断结果,良性或恶性。这是我们要预测的结果变量。
研究者对9个特征进行了评分和编码,评分的范围是1~10。可以在R语言的MASS包中找到该数据框,名为 biopsy。为进行数据准备,我们加载这个数据框,确认数据结构,将变量重命名为有意义的名称,并删除缺失值。代码如下所示:
library(MASS)
data(biopsy)
str(biopsy)
## 'data.frame': 699 obs. of 11 variables:
## $ ID : chr "1000025" "1002945" "1015425" "1016277" ...
## $ V1 : int 5 5 3 6 4 8 1 2 2 4 ...
## $ V2 : int 1 4 1 8 1 10 1 1 1 2 ...
## $ V3 : int 1 4 1 8 1 10 1 2 1 1 ...
## $ V4 : int 1 5 1 1 3 8 1 1 1 1 ...
## $ V5 : int 2 7 2 3 2 7 2 2 2 2 ...
## $ V6 : int 1 10 2 4 1 10 10 1 1 1 ...
## $ V7 : int 3 3 3 3 3 9 3 3 1 2 ...
## $ V8 : int 1 2 1 7 1 7 1 1 1 1 ...
## $ V9 : int 1 1 1 1 1 1 1 1 5 1 ...
## $ class: Factor w/ 2 levels "benign","malignant": 1 1 1 1 1 2 1 1 1 1 ...
对数据结构的检查发现,特征变量均是整数型变量,结果变量是因子变量,因此不需要将数据转换为其他结构。我们可以删除ID列,如下所示:
biopsy$ID = NULL
接着重命名变量,并确认操作结果:
names(biopsy) = c("thick", "u.size", "u.shape", "adhsn",
"s.size", "nucl", "chrom", "n.nuc", "mit", "class")
names(biopsy)
## [1] "thick" "u.size" "u.shape" "adhsn" "s.size" "nucl" "chrom"
## [8] "n.nuc" "mit" "class"
接下来,我们删除那些有缺失值的观测。因为只有16个观测有缺失数据,
只占所有观测的2%,
所以删除它们问题不大。通过删除这些观测,我们建立了一个新的工作数据框。使用na.omit()函数后,一行代码就可以删除所有有缺失数据的观测。
biopsy.v2
根据你分析数据时使用的R包的不同,结果变量也可能要求是数值型,比如0或者1。为了满足这个要求,可以创建一个变量y,用0表示良性,用1表示恶性,然后使用ifelse()函数为y赋值,如下所示:
y "malignant", 1, 0)
数据准备的最终任务是建立训练数据集和测试数据集。从原始数据中建立两个不同的数据集的目的在于,这样可以更准确地预测未使用数据或未知数据。实际上,在机器学习中,我们不应该过于关心对已使用的观测的预测有多好,而应该把重点放在那些建立算法时没有使用过的观测的预测上面。所以,我们要使用训练数据建立并选择一个对测试数据能做出最好预测的最优算法。在本章中,我们会遵循这个评价标准来建立模型。有多种方式可以将数据恰当地划分成训练集和测试集:50/50、60/40、70/30、80/20,诸如此类。你应该根据自己的经验和判断选择数据划分方式。在本例中,我选择按照70/30的比例划分数据。如下所示:
set.seed(123
) #random number generator
ind 2, nrow(biopsy.v2), replace = TRUE, prob = c(0.7, 0.3))
train 1, ] #the training data set
test 2, ] #the test data setstr(test) #confirm it worked
## 'data.frame': 209 obs. of 10 variables:
## $ thick : int 5 6 4 2 1 7 6 7 1 3 ...
## $ u.size : int 4 8 1 1 1 4 1 3 1 2 ...
## $ u.shape: int 4 8 1 2 1 6 1 2 1 1 ...
## $ adhsn : int 5 1 3 1 1 4 1 10 1 1 ...
## $ s.size : int 7 3 2 2 1 6 2 5 2 1 ...
## $ nucl : int 10 4 1 1 1 1 1 10 1 1 ...
## $ chrom : int 3 3 3 3 3 4 3 5 3 2 ...
## $ n.nuc : int 2 7 1 1 1 3 1 4 1 1 ...
## $ mit : int 1 1 1 1 1 1 1 4 1 1 ...
## $ class : Factor w/ 2 levels "benign","malignant": 1 1 1 1 1 2 1 2 1 1 ...
## - attr(*, "na.action")= 'omit' Named int 24 41 140 146 159 165 236 250 276 293 ...
## ..- attr(*, "names")= chr "24" "41" "140" "146" ...
为了确保两个数据集的结果变量是均衡的,我们做以下检查:
table(train$class)
##
## benign malignant
## 302 172
table(test$class)
##
## benign malignant
## 142 67
两个数据集的结果变量的内部比例完全可以接受,既然如此,下面开始进行模型构建和模型评价。
3. 模型构建与模型评价
我们首先用所有自变量建立一个Logistic回归模型,然后用最优子集法对特征进行削减。在此之后,我们会试验判别分析和多元自适应回归样条(MARS)的方法。
R中的glm()函数可以拟合广义线性模型,其中包括
Logistic回归
模型,我们必须在函数中使用family = binomial这个参数。该参数告诉R运行
Logistic回归
,而不是其他版本的广义线性模型。首先在训练数据集上使用所有特征建立一个模型,看看这个模型在测试数据集上运行的效果。如下所示:
full.fit
##
## Call:
## glm(formula = class ~ ., family = binomial, data = train)
##
## Deviance Residuals:
## Min 1Q Median 3Q Max
## -3.3397 -0.1387 -0.0716 0.0321 2.3559
##
## Coefficients:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) -9.4293 1.2273 -7.683 1.55e-14 ***
## thick 0.5252 0.1601 3.280 0.001039 **
## u.size -0.1045 0.2446 -0.427 0.669165
## u.shape 0.2798 0.2526 1.108 0.268044
## adhsn 0.3086 0.1738 1.776 0.075722 .
## s.size 0.2866 0.2074 1.382 0.167021
## nucl 0.4057 0.1213 3.344 0.000826 ***
## chrom 0.2737 0.2174 1.259 0.208006
## n.nuc 0.2244 0.1373 1.635 0.102126
## mit 0.4296 0.3393 1.266 0.205402
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## (Dispersion parameter for binomial family taken to be 1)
##
## Null deviance: 620.989 on 473 degrees of freedom
## Residual deviance: 78.373 on 464 degrees of freedom
## AIC: 98.373
##
## Number of Fisher Scoring iterations: 8
通过summary()函数,我们可以查看各个预测变量的系数及其p值。可以看到,只有两个特征的p值小于0.05(thickness和nuclei)。使用confint()函数可以对模型进行95%置信区间的检验。如下所示:
confint(full.fit)
## Waiting for profiling to be done...
## 2.5 % 97.5 %
## (Intercept) -12.23786660 -7.3421509
## thick 0.23250518 0.8712407
## u.size -0.56108960 0.4212527
## u.shape -0.24551513 0.7725505
## adhsn -0.02257952 0.6760586
## s.size -0.11769714 0.7024139
## nucl 0.17687420 0.6582354
## chrom -0.13992177 0.7232904
## n.nuc -0.03813490 0.5110293
## mit -0.14099177 1.0142786
对数函数log(P(Y)/1-P(Y))=B0+B1x的B系数可以通过exponent(beta)指数函数转化为优势比。要在R中计算优势比,可以使用下面的exp(coef())函数形式:
exp(coef(full.fit))
## (Intercept) thick u.size u.shape adhsn
## 8.033466e-05 1.690879e+00 9.007478e-01 1.322844e+00 1.361533e+00
## s.size nucl chrom n.nuc mit
## 1.331940e+00 1.500309e+00 1.314783e+00 1.251551e+00 1.536709e+00
优势比可以解释为特征中1个单位的变化导致的结果发生比的变化。如果系数大于1,就说明当特征的值增加时,结果的发生比会增加。反之,系数小于1就说明,当特征的值增加时,结果的发生比会减小。本例中,除u.size之外的所有特征都会增加对数发生比。
我们进行数据探索时发现一个问题:潜在的多重共线性可能。同线性回归一样,在Logistic回 归中也可以算出方差膨胀因子统计量(
VIF
),如下所示:
library(car)
## Loading required package: carData
vif(full.fit)
## thick u.size u.shape adhsn s.size nucl chrom n.nuc
## 1.235204 3.248811 2.830353 1.302178 1.635668 1.372931 1.523493 1.343145
## mit
## 1.059707
没有一个VIF值大于5,根据VIF经验判断法则,共线性并不明显。下面的任务就是进行变量(机器学习中一般称作"特征")选择。我们先看看模型在训练数据集和测试数据集上表现如何。
首先要建立一个向量,表示预测概率,如下所示:
train.probs "response")
train.probs[1:5] #inspect the first 5 predicted probabilities
## 1 3 6 7 9
## 0.02052820 0.01087838 0.99992668 0.08987453 0.01379266
contrasts(train$class)
## malignant
## benign 0
## malignant 1
下一步需要评价模型在训练集上预测的效果,然后再评价它在测试集上的拟合程度。快速实现评价的方法是生成一个混淆矩阵。InformationValue包可以实现混淆矩阵。这时,我们需要用0和1来表示结果。函数区别良性结果和恶性结果使用的默认值是0.50,也就是说,当概率大于等于0.50时,就认为这个结果是恶性的:
library(InformationValue)
trainY 1]
testY 2]
confusionMatrix(trainY, train.probs)
## 0 1
## 0 294 7
## 1 8 165
矩阵的行表示预测值,列表示实际值。对角线上的元素是预测正确的分类。右上角的值7表示误诊为恶性的预测数,左下角的值8表示误诊为良性的预测数。可以查看错误预测的比例。如下所示:
misClassError(trainY, train.probs)
## [1] 0.0316
训练集上只有3.16%的预测错误率。如前所述,我们必须正确预测未知数据,换句话说,必须正确预测测试集。在测试集上建立混淆矩阵的方法和在训练集上一样:
test.probs "response")
misClassError(testY, test.probs)
## [1] 0.0239
confusionMatrix(testY, test.probs)
## 0 1
## 0 139 2
## 1 3 65
我们使用全部特征建立的模型预测效果很好,约98%的预测正确率。但是,我们还是要看看是否有改进的空间。
交叉验证的目的是提高测试集上的预测正确率,以及尽可能避免过拟合。K折交叉验证的做法是将数据集分成K个相等的等份,每个等份称为一个K子集(K-set)。算法每次留出一个子集,使用其余K-1个子集拟合模型,然后用模型在留出的那个子集上做预测。将上面K次验证的结果进行平均,可以使误差最小化,并且获得合适的特征选择。你也可以使用留一交叉验证方法,这里的K等于N。模拟表明,LOOCV可以获得近乎无偏的估计,但是会有很高的方差。所以,
大多数机器学习专家都建议将K的值定为5或10
。
bestglm包可以自动进行交叉验证,这个包依赖于我们在线性回归中使用过的leaps包。
library(bestglm)
## Loading required package: leaps
加载程序包之后,需要将结果编码成0或1。如果结果仍为因子,程序将不起作用。使用这个程序包的另外一个要求是结果变量(或y)必须是最后一列,而且要删除所有没有用的列。通过以下代码新建一个数据框即可快速实现上述要求:
X 1:9]
Xy "CV", CVArgs = list(Method = "HTF", K = 10, REP = 1),
family=binomial)
## Morgan-Tatar search since family is non-gaussian.
## CV(K = 10, REP = 1)
## BICq equivalent for q in (7.16797006619085e-05, 0.273173435514231)
## Best Model:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) -7.8147191 0.90996494 -8.587934 8.854687e-18
## thick 0.6188466 0.14713075 4.206100 2.598159e-05
## u.size 0.6582015 0.15295415 4.303260 1.683031e-05
## nucl 0.5725902 0.09922549 5.770596 7.899178e-09
在上面的代码中,Xy = Xy指的是我们已经格式化的数据框,IC = “CV”告诉程序使用的信息准则为交叉验证,CVArgs是我们要使用的交叉验证参数。HTF方法就是K折交叉验证,后面的数字K = 10指定了均分的份数,参数REP = 1告诉程序随机使用等份并且只迭代一次。像glm()函数一样,我们需要指定参数family = binomial。另外,如果指定参数family = gaussian,你就可以使用bestglm()函数进行线性回归。完成上面的交叉验证分析之后,会得到下面的结果:Best Model有三个特征:thick、u.size和nucl。Morgan-Tatar搜索的结果表明,程序对所有可能的子集进行了简单而彻底的搜索。
我们可以把这些特征放到glm()函数中,看看模型在测试集上表现如何。predict()函数不能用于bestglm生成的模型,所以下面的步骤是必需的:
reduce.fit
同前面一样,下面的代码可以在测试集上比较预测值和实际值:
test.cv.probs = predict(reduce.fit, newdata = test, type = "response")
misClassError(testY, test.cv.probs)
## [1] 0.0383
confusionMatrix(testY, test.cv.probs)
## 0 1
## 0 139 5
## 1 3 62
精简了特征的模型和全特征模型full.fit相比,精确度略有下降。我们可以用bestglm包再试一次,这次使用信息准则为BIC的最优子集方法:
bestglm(Xy = Xy, IC = "BIC", family = binomial)
## Morgan-Tatar search since family is non-gaussian.
## BIC
## BICq equivalent for q in (0.273173435514231, 0.577036596263757)
## Best Model:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) -8.6169613 1.03155250 -8.353391 6.633065e-17
## thick 0.7113613 0.14751510 4.822295 1.419160e-06
## adhsn 0.4537948 0.15034294 3.018398 2.541153e-03
## nucl 0.5579922 0.09848156 5.665956 1.462068e-08
## n.nuc 0.4290854 0.11845720 3.622282 2.920152e-04
试过所有可能的子集之后,以上4个特征提供了最小的BIC评分。我们看看这个模型在测试集上的预测效果,如下所示:
bic.fit "response")
misClassError(testY, test.bic.probs)
## [1] 0.0239
confusionMatrix(testY, test.bic.probs)
## 0 1
## 0 138 1
## 1 4 66
我们得到5个错误的预测,和全特征模型一样。那么问题来了:哪一个模型更好?在任何正常情况下,如果效果差不多,经验法则会选择最简单的和/或最好解释的模型。我们当然可以开启一个全新的分析,使用新的随机数以及新的训练集和测试集划分比例。但是,不妨假定我们已经做了Logistic回归能做的所有操作,最后还是回到了全特征模型和BIC最小模型,仍然需要讨论模型选择的方法。下面讨论判别分析的方法,在最后的模型推荐中,我们有可能选择使用这种方法建立的模型。
4. 判别分析应用
当分类很确定时,判别分析可以有效替代Logistic回归。当你遇到分类结果很确定的分类问题时,
Logistic回归
的估计结果可能是不稳定的,即置信区间很宽,不同样本之间的估计值会有很大变化(James,2013)。判别分析不会受到这个问题的困扰,实际上,它会比
Logistic回归
做得更好。反之,如果特征和结果变量之间具有错综复杂的关系,判别分析在分类任务上的表现就会非常差。在上述乳腺癌活检数据这个案例中,
Logistic回归
在训练集和测试集上的表现都非常好,分类的结果并不确定。出于同逻辑回归进行比较的目的,我们研究一下判别分析,包括线性判别分析和二次判别分析。
判别分析使用贝叶斯定理确定每个观测属于某个类别的概率。如果你有两个类别,比如良性和恶性,判别分析会计算观测分别属于两个类别的概率,然后选择高概率的类别作为正确的类别。贝叶斯定理定义了在X已经发生的条件下Y发生的概率:等于Y和X同时发生的概率除以X发生的概率,公式如下:
Y/X的概率= P(X+Y)/P(X)
公式中的分子表示一个具有某些特征的观测属于某个分类水平的可能性,分母表示一个具有这些特征的观测属于所有分类水平的可能性。同样地,分类原则认为如果X和Y的联合分布已知,那么给定X后,决定观测属于哪个类别的最佳决策是选择那个有更大概率(后验概率)的类别。获得后验概率的过程如下所示。
(1)收集已知类别的数据。
(2)计算先验概率——代表属于某个类别的样本的比例。
(3)按类别计算每个特征的均值。
(4)计算每个特征的方差协方差矩阵。在线性判别分析中,这会是一个所有类别的混合(5)矩阵, 给出线性分类器;在二次判别分析中,会对每个分类建立一个方差协方差矩阵。
(6)估计每个分类的正态分布(高斯密度)。
(7)计算discriminant函数,作为一个新对象的分类原则。
(8)根据discriminant函数,将观测分配到某个分类。
线性判别分析假设每种类别中的观测服从多元正态分布,并且不同类别之间的具有同样的协方差。二次判别分析仍然假设观测服从正态分布,但假设每种类别都有自己的协方差。重要的是要记住,
二次判别分析技术比Logistic回归更具灵活性
,同时还要时刻牢记偏差/方差权衡的问题。使用更有灵活性的技术可以得到偏差更小的结果,但很可能具有更高的方差。和很多灵活的技术一样,需要一个高鲁棒性的训练数据集来降低高分类方差。
线性判别分析(LDA)可以用MASS包实现
,我们为了使用biopsy数据集已经加载了这个包。LDA的语法和lm()以及glm()函数非常相似。LDA模型的拟合如下所示:
lda.fit
## Call:
## lda(class ~ ., data = train)
##
## Prior probabilities of groups:
## benign malignant
## 0.6371308 0.3628692
##
## Group means:
## thick u.size u.shape adhsn s.size nucl chrom
## benign 2.92053 1.304636 1.413907 1.324503 2.115894 1.397351 2.082781
## malignant 7.19186 6.697674 6.686047 5.668605 5.500000 7.674419 5.959302
## n.nuc mit
## benign 1.225166 1.092715
## malignant 5.906977 2.639535
##
## Coefficients of linear discriminants:
## LD1
## thick 0.19557291
## u.size 0.10555201
## u.shape 0.06327200
## adhsn 0.04752757
## s.size 0.10678521
## nucl 0.26196145
## chrom 0.08102965
## n.nuc 0.11691054
## mit -0.01665454
从结果可以看出,在分组先验概率中,良性概率大约为64%,恶性概率大约为36%。下面再看看分组均值,这是按类别分组的每个特征的均值。线性判别系数是标准线性组合,用来确定观测的判别评分的特征。评分越高,越可能被分入恶性组。对LDA模型使用plot()函数,可以画出判别评分的直方图和密度图,如下所示:
plot(lda.fit, type="both")
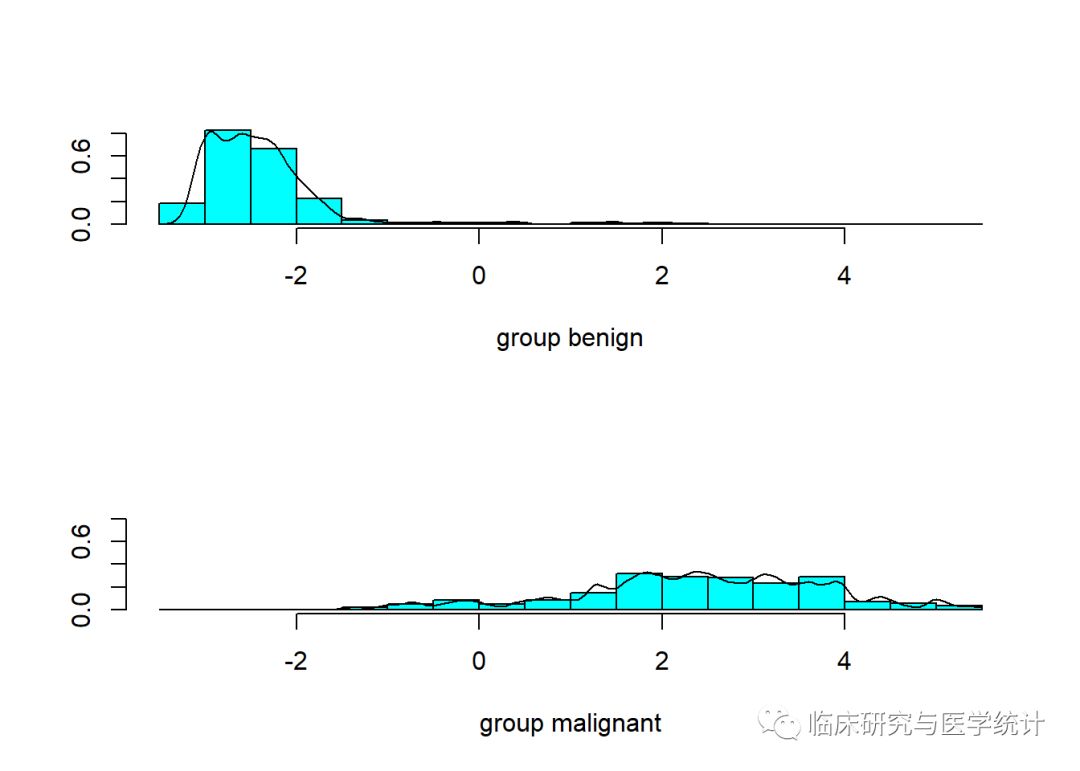
可以看出,组间有些重合,这表明有些观测被错误分类。LDA模型可以用predict()函数得到3种元素(class、posterior和x)的列表。class元素是对良性或恶性的预测,posterior是值为x的评分可能属于某个类别的概率,x是线性判别评分。通过下面的函数,我们仅提取恶性观测的概率:
train.lda.probs 2]
misClassError(trainY, train.lda.probs)
## [1] 0.0401
confusionMatrix(trainY, train.lda.probs)
## 0 1
## 0 296 13
## 1 6 159
LDA模型在训练集上表现得比Logistic回归模型差多了。但最重要的是LDA模型在测试集上的表现,如下所示:
test.lda.probs 2]
misClassError(testY, test.lda.probs)
## [1] 0.0383
confusionMatrix(testY, test.lda.probs)
## 0 1
## 0 140 6
## 1 2 61
它在测试集上的表现比我预期的要好。从正确分类率的角度看,LDA模型表现得依然不如Logistic回归模型(LDA模型:96%,
Logistic回归
模型:98%)。下面用二次判别分析(QDA)模型来拟合数据。
R中QDA也是MASS包的一部分,函数为qda()。
我们将模型存储在一个名为qda.fit的对象中,如下所示:
qda.fit
## Call:
## qda(class ~ ., data = train)
##
## Prior probabilities of groups:
## benign malignant
## 0.6371308 0.3628692
##
## Group means:
## thick u.size u.shape adhsn s.size nucl chrom
## benign 2.92053 1.304636 1.413907 1.324503 2.115894 1.397351 2.082781
## malignant 7.19186 6.697674 6.686047 5.668605 5.500000 7.674419 5.959302
## n.nuc mit
## benign 1.225166 1.092715
## malignant 5.906977 2.639535
结果中有分组均值,和LDA一样;但是没有系数,因为这是二次函数,我们前面讨论过了。使用与LDA相同的代码,可以得到QDA模型在训练集和测试集上的预测结果:
train.qda.probs 2]
misClassError(trainY, train.qda.probs)
## [1] 0.0422
confusionMatrix(trainY, train.qda.probs)
## 0 1
## 0 287 5
## 1 15 167
test.qda.probs 2]
misClassError(testY, test.qda.probs)
## [1] 0.0526
confusionMatrix(testY, test.qda.probs)
## 0 1
## 0 132 1
## 1 10 66
根据混淆矩阵可以立即断定,QDA模型在训练数据集上表现得最差。QDA在测试集上的分类效果也很差,有11个预测错误,而误为恶性的比例尤其高。
5. 多元自适应回归样条方法
如果有一种建模技术具有以下特点,应该会很受欢迎:
(1)对于回归和分类问题,都可以灵活建立线性模型和非线性模型。
(2)支持变量的交互项。
(3)简单易懂,易于解释。
(4)几乎不需要数据预处理。
(5)可以处理所有类型的数据:数值型、因子等。
(6)可以很好地预测未知数据,也就是说,在偏差/方差的权衡方面做得非常好。
MARS可以非常容易做到这些。首先,我们从一个前面已经讨论过的线性模型或广义线性模型开始。然后,为了捕获非线性关系,添加一个铰链(hinge)函数。这些铰链作用于输入特征,相当于系数的改变。举例来说,假设有这样一个模型:Y=12.5(截距)+1.5(变量1)+3.3(变量2),其中变量1和变量2的范围都在1和10之间。下面,我们看看变量2的铰链函数如何发挥作用:
Y = 11(新截距)+1.5(变量1)+ 4.26734(max(0, 变量2 - 5.5))
于是,我们可以这样理解铰链函数:在0和变量2减去5.50的结果中,它找出其中的最大值。所以,只要变量2的值大于5.5,铰链函数的值就会乘以系数,否则它的值就是0。这种方法可以给每个变量配置多个铰链,也可以用于交互项。MARS方法的另一个有趣之处是,它可以自动进行变量选择。这是通过交叉验证实现的,默认的方法是通过前向过程建模——这非常类似于前向逐步回归——然后通过后向过程精简模型。因为模型完成前向过程之后,有可能出现对数据的过拟合,所以要通过后向过程根据广义交叉验证对输入特征进行精简,并去除铰链。
GCV=RSS/N*(1 - 参数有效数量/N)^2
参数有效数量 = 输入特征数量 + 惩罚系数*(输入特征数量 - 1)/2
在earth包中,对于加法模型来说,惩罚系数 = 2;对于乘法模型,其中有交互项,惩罚系数 = 3。
R中可以调整的参数非常少。在下面例子中,演示一种简单而有效实现MARS的方法。你可以使用MDA包,也可以通过earth包实现。代码和前面的例子很相似,在那里我们使用了glm()函数。但需要注意,一定要设定如何精简模型,并且使响应变量服从二元分布。我的设定是使用5折交叉验证来选择模型(pmethod=“cv”,nfold=5),重复3次(ncross=3);使用没有交互项的加法模型(degree=1),每个输入特征只使用一个铰链函数(minspan=1)。在我使用的数据中,交互项和多个铰链函数都会导致过拟合。当然,你的情况会有所不同。代码如下所示:
library(earth)
## Loading required package: plotmo
## Loading required package: plotrix
## Loading required package: TeachingDemos
set.seed(1)
earth.fit "cv",
nfold = 5,
ncross = 3,
degree = 1,
minspan = -1,
glm=list(family=binomial)
)
## Warning: glm.fit: algorithm did not converge
## Warning: glm.fit: fitted probabilities numerically 0 or 1 occurred
## Warning: glm.fit: algorithm did not converge
## Warning: glm.fit: fitted probabilities numerically 0 or 1 occurred
下面查看模型摘要。我们可以看到模型公式和Logistic回归系数,还有铰链函数,再后面是一些注释和广义R方的数值,等等。MARS模型的生成过程是:先根据数据建立一个标准线性回归模型,它的响应变量在内部编码为0和1;对特征(变量)进行精简之后,生成最终模型,并将其转换为一个GLM模型。所以,我们可以不用考虑R方的值:
summary(earth.fit)
## Call: earth(formula=class~., data=train, pmethod="cv",
## glm=list(family=binomial), degree=1, nfold=5, ncross=3,
## minspan=-1)
##
## GLM coefficients
## malignant
## (Intercept) -6.5746417
## u.size 0.1502747
## adhsn 0.3058496
## s.size 0.3188098
## nucl 0.4426061
## n.nuc 0.2307595
## h(thick-3) 0.7019053
## h(3-chrom) -0.6927319
##
## Earth selected 8 of 10 terms, and 7 of 9 predictors using pmethod="cv"
## Termination condition: RSq changed by less than 0.001 at 10 terms
## Importance: nucl, u.size, thick, n.nuc, chrom, s.size, adhsn, ...
## Number of terms at each degree of interaction: 1 7 (additive model)
## Earth GRSq 0.8354593 RSq 0.8450554 mean.oof.RSq 0.8331308 (sd 0.0295)
##
## GLM null.deviance 620.9885 (473 dof) deviance 81.90976 (466 dof) iters 8
##
## pmethod="backward" would have selected the same model:
## 8 terms 7 preds, GRSq 0.8354593 RSq 0.8450554 mean.oof.RSq 0.8331308
模型有8项,包括截距和7个预测变量。其中两个预测变量有铰链函数,这就是浓度和染色质变量。如果浓度大于3,就会用系数0.7019乘以铰链函数的值,否则这一项就是0。对于染色质,如果它的值小于3,那么就用系数乘以铰链函数值,否则这一项就是0。还可以做出统计图。第一张图是用plotmo()函数生成的,它展示了保持其他预测变量不变,某个预测变量发生变化时,响应变量发生的改变。你可以清楚地看到铰链函数对浓度所起的作用:
plotmo(earth.fit)
## plotmo grid: thick u.size u.shape adhsn s.size nucl chrom n.nuc mit
## 4 1 2 1 2 1 3 1 1

通过plotd()函数,可以生成按类别标签分类的预测概率密度图:
plotd(earth.fit)
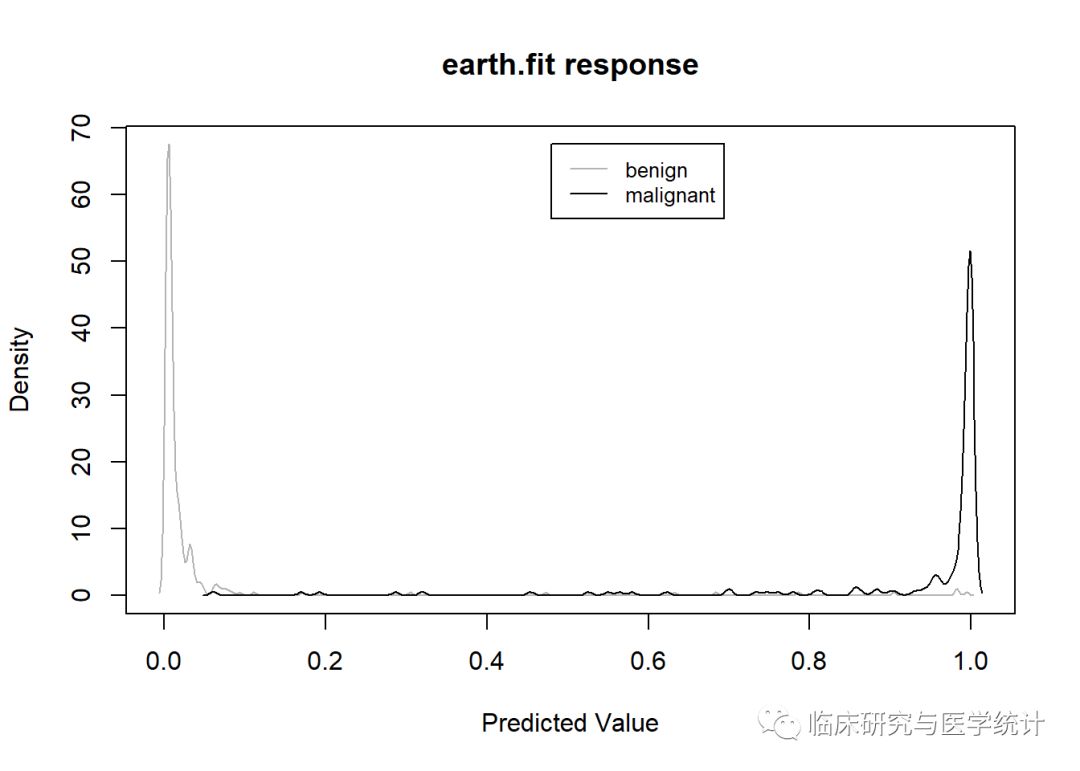
下面看看变量之间的相对重要性。先解释一下,nsubsets是精简过程完成之后包含这个变量的模型的个数。对于gcv和rss列,其中的值表示这个变量贡献的gcv和rss值的减少量(gcv和rss的范围都是0~100):
evimp(earth.fit)
## nsubsets gcv rss
## nucl 7 100.0 100.0
## u.size 6 44.2 44.8
## thick 5 23.8 25.1
## n.nuc 4 15.1 16.8
## chrom 3 8.3 10.7
## s.size 2 6.0 8.1
## adhsn 1 2.3 4.6
我们再看一下模型在测试集上的表现:
test.earth.probs "response")
misClassError(testY, test.earth.probs)
## [1] 0.0287
confusionMatrix(testY, test.earth.probs)
## 0 1
## 0 138 2
## 1 4 65
这几乎可以和Logistic回归模型相比。