▎药明
康德/报道
TP53
基因是人类癌症中最常出现突变的抑癌基因。
p53蛋白能够被多种应激刺激激活,同时它也控制着一个非常复杂的抗增殖转录系统,这一转录系统影响到的生物反应多得让人看着头晕。虽然我们已经揭示了p53信号网络的许多方面,但是对p53在什么情况下如何行使它的多种功能还没有清晰的了解。细胞种类,基因突变形态和细胞状态是如何影响p53的功能的?我们怎样才能在癌症中恢复它抑制肿瘤的活性?日前《细胞》杂志对p53进行了深度盘点。
p53是在肿瘤病毒研究的巅峰时期被发现的,最初它被认为是致癌基因,但是随后的研究发现野生型(wild type) p53在细胞培养环境下抑制肿瘤生长和细胞癌变。在编码p53的
TP53
基因中出现的失活性突变在人类肿瘤中非常常见。在很多癌症中,
TP53
基因突变与患者的不良预后相关。
p53是一种调控转录的序列特异性DNA结合蛋白。
与p53介导的转录调控在抑制肿瘤中的重要作用一致,大部分肿瘤中出现的
TP53
基因突变发生在编码p53蛋白DNA结合域的部分。
在正常细胞中,p53被一系列调节因子维持在低水平。
其中
MDM2
基因作为p53泛素连接酶,能够促进p53的降解。不过在多种细胞应激情况下p53蛋白在细胞中的稳定性得到提高。激活p53的机制根据刺激的不同而改变,例如,DNA损伤会促进p53的磷酸化,从而阻断
MDM2
媒介的降解。而致癌基因突变引发的信号通路会诱导ARF肿瘤抑制子抑制
MDM2
的功能。
p53的功能中了解最为深入的是它促进细胞周期停滞(cell cycle arrest)和细胞凋亡(apoptosis)的功能。
90年代早期的重要研究表明p53对媒介可逆DNA损伤引发G1期检查点的激活非常重要,它促进细胞周期停滞的目的应该是为了帮助DNA在进行更多细胞分裂之前获得修复。同时,p53可以通过引导促进凋亡的BCL-2家族成员的表达来促使细胞凋亡的发生。
为什么p53是一个这么重要的肿瘤抑制子呢?
一方面,p53在DNA出现损伤后能够引发细胞周期停滞或者消灭细胞,表明它可以通过预防致癌基因突变的累积来防止癌症发生。从这个角度来说,
p53缺失会导致存活下来的子细胞中基因突变数目增多,从而间接地促进癌症的发生
。另一方面,p53在致癌基因异常表达时能够阻止细胞增殖,表明它可能在限制致癌基因突变的恶果方面也有重要作用。在这种情况下,
p53缺失会允许表达致癌基因的细胞继续无限制增殖,从而直接导致癌症发生。
不管从哪个角度来说,p53都承担着基因组守护者(guardian of the genome)的角色,它会限制基因突变带来的有害后果。
虽然这种理解能够为解释“为什么
TP53
基因突变在人类癌症中最为常见?”提供了一个基本框架,但是最近的研究为p53的功能做出了更为细致入微的描述。这些研究表明p53激活后的结果非常广泛多样,而且它对基因的转录调控与很多背景因素相关。
当DNA损伤发生后,DNA 损伤反应(DNA damage response, DDR) 激酶会磷酸化p53,激活p53,导致细胞周期停滞、细胞衰老(senescence)或者细胞凋亡。
这些步骤最终可以减少基因突变进一步传播的风险。
而且,p53会激活编码DNA修复机制组成部分的基因,从而刺激DNA修复。
因此
TP53
基因突变经常与单核苷酸变异(single nucleotide variants, SNVs) 和特定的协同变异(co-mutated)基因相关联,这并不令人惊讶。
令人惊讶的是在肿瘤分析中
TP53
基因突变与基因拷贝数变异(copy number variation, CNV)有非常紧密的联系。
而且,携带
TP53
基因变异的肿瘤细胞通常为非整倍体(aneuploid),在完整染色体的数目上出现明显变化。
这种情况出现的原因是多方面的,
其中一个原因与p53调节G2/M细胞周期转换的能力有关。
例如,p53缺失导致对MAD2的抑制减弱,从而导致纺锤体组装检查点(spindle assembly checkpoint)失调。这会引起染色体出现错误分离(missegregation)和四倍体的机率上升。在四倍体细胞中,p53缺失会导致多极有丝分裂的几率增加,从而加剧染色体错误分离。
另一个原因是p53可以通过杀死可能出现异常有丝分裂的细胞来限制染色体不稳定性的产生,特别是在中心体扩增或端粒功能失常之后。
多余的中心体可以导致Hippo信号通路的上调,这会通过抑制MDM2来激活p53。有研究表明p53缺失的细胞更容易耐受异常基因剂量造成的蛋白组方面的压力。综合这些原因,p53缺失似乎不但促进非整倍体细胞的产生,而且维持它们的存活。
另一个p53维持基因组完整性的手段是抑制反转录转座子(retrotransposon)的移动。
反转录转座子是病毒生成的遗传元件,它们可能通过在基因组中的转移和重新插入造成基因突变的产生。有研究表明在果蝇中激活可移动的反转录转座子可以导致DNA出现双链断裂,p53引发的细胞凋亡理论上可以减少DNA双链断裂带来的基因突变的可能。但是p53基因突变与反转录转座子表达之间的关系并不只限于消灭出错细胞。p53可以与LINE元件(LINE elements)和其它转座子内的序列相结合并且导致它们的表达水平下降。p53对反转录转座子的抑制依靠的是在表观遗传层面对反转录转座子位点的抑制,而不是细胞凋亡。如果对反转录转座子的抑制减弱,它们可以重新整合到基因组中并且增加基因突变的产生。
虽然基因组不稳定性不是肿瘤发生的必须条件,但是p53缺失导致的基因组不稳定性让肿瘤细胞可能积累更多致癌驱动子,从而加速癌变、肿瘤转移和抗药性的产生。对于一个肿瘤来说,它包含的肿瘤细胞中基因突变的多样性,而不是基因突变的绝对数目,决定了肿瘤细胞群体在面临不同环境和挑战时表现出的韧性。从这个角度来说,
p53失活的独特之处在于它不但促进基因组不稳定性的产生(提高新基因突变产生的频率),而且维持携带广泛遗传错误的细胞存活(减少基因突变消失的几率)。
这些研究表明p53失活可能帮助建立肿瘤内异质性(intratumoral heterogeneity)。
调节基因组完整性,细胞周期停滞和细胞凋亡,似乎这些对一个基因而言还不够多似的,日益增多的研究表明,
p53同时还控制更多“非经典”通路
。例如,p53可以调节自噬活动,变动新陈代谢,抑制多能性和细胞可塑性,并且协助一种称为铁死亡(ferrotoposis)的依靠铁离子的细胞死亡方式。即便是基准水平的p53也可以增强多种其它肿瘤抑制信号网络。然而,
令人惊讶的是这么多研究并没有给我们一个简单而明确的答案指出p53的功能到底是什么,以及它是怎样完成这些功能的。
我们目前得到的结论是p53造成的生物反应异乎寻常的灵活,它会因为细胞种类、细胞分化状态、应激条件和环境中的配合信号的改变而改变。
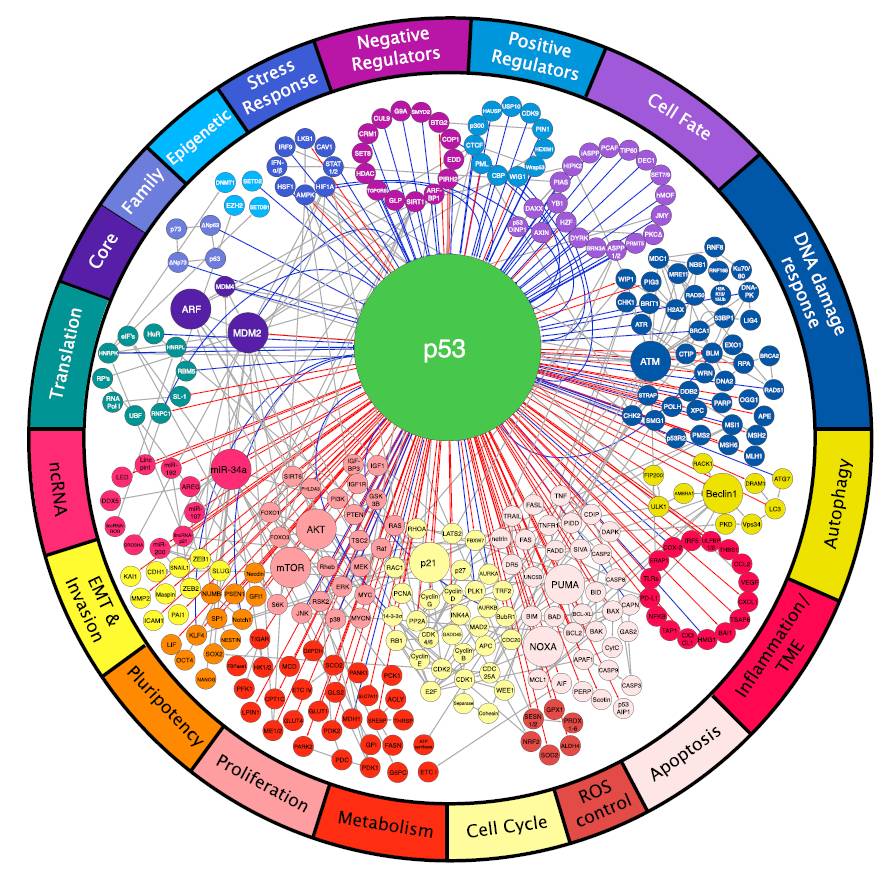
▲p53信号网络
p53多种多样的功能来源于它在不同条件下可以调控不同集合的靶向基因的能力。
试图发现一组通用的p53靶向基因的研究都不可避免地遭受失败。
对16组全基因组数据进行的荟萃分析(meta-analysis)只发现大约60个基因是共同靶点。这些研究发现的中心准则是,细胞环境和刺激的不同会导致功能上不同组的基因被激活,而不仅仅是同一组基因的激活水平不同。我们不能指望在不同组织中致癌基因的活性(例如
KRAS
会在结肠、胰腺和肺组织中被激活)会导致同样p53调控的转录反应。而且,我们也无法假设在单细胞水平上DNA损伤激活p53造成的基因表达特征,会和致癌基因激发的基因表达特征相同。是什么原因导致p53调控的多种信号通路在特定生理条件下只有一种或少数几种被激活,仍然需要更多系统性的研究。
细胞新陈代谢是最近获得关注的一条p53调控的非经典生理过程。
p53调控的基因可以调控包括谷氨酰胺分解代谢,抗氧化活性,脂类合成,脂肪酸氧化和糖异生等代谢过程。而且根据细胞种类的不同,p53可以对同一代谢过程施加相反的作用。例如,在乳腺和肺癌细胞中,p53通过减弱葡萄糖吸收或者抑制糖酵解酶的表达来达到抑制糖酵解(glycolysis)的作用。然而在肌肉细胞中,p53通常引导糖酵解酶的表达。这些研究结果意味着p53能够调节新陈代谢的不同方面,从而产生不同、甚至是相反的生化和表型反应。
而p53引发不同生物反应的原理仍然不明。
一方面,p53能够依据细胞种类和刺激类型来引导功能上不同的转录程序,从而产生不同的生物后果。目前提出的一个p53调节不同生物反应的机制是通过基于刺激的翻译后修饰(post-translational modification, PTMs)。对p53的PTMs能够改变p53对不同靶点基因的亲合力。例如,磷酸化p53(S46)和乙酰化p53(K120)可以激发细胞凋亡,而PRMT5-甲基化p53相比之下则更容易激活p21。另外在p53蛋白中出现的诸如SUMO化(SUMOylation)、糖基化(glycosylation)和脯氨酰异构化(prolyl isomerization)等PTMs可以改变蛋白的稳定性,并且影响对靶点基因的偏好。
诱发p53表达可以导致稳定的信号输出或者震荡式的波动信号。
不同寻常的是,p53表达变化的动力学特征,可以独立于最高p53蛋白水平,决定细胞在面对遗传毒性压力(genotoxic stress)时的反应。p53激活时的动力学特征之所以能够转化为对不同靶点的倾向是因为p53对不同靶点的结合和分离速率不一样。例如,p21的启动子对短暂的p53脉冲更为敏感,而p53的另一个靶点,促凋亡的FAS则对这种p53信号不敏感。因此,短暂的p53脉冲会导致增殖停滞,而持续信号则引发细胞凋亡。
另一方面,多个因素影响细胞如何解释p53的激活。
第一,细胞谱系(cell lineage)可能是决定细胞在多种不同反应中倾向哪一种的重要决定因素。比如,细胞种类和细胞状态决定的染色质修饰可能导致某些基因更容易或更难与激活的p53结合。例如,在胚胎干细胞中,p53可以被激活并且与p21启动子结合,但是p21是否会被有效地激活取决于在这一位点上基于细胞种类的抑制性H3K27me3修饰。第二,p53靶向的基因图谱可以被其它合作或对抗的转录因子影响,它们包括FOXO和NF-B。最后,同样的转录信号输出可能由于细胞状态的不同而导致不同的结果。比如ATM信号通路可以保护细胞不发生p53媒介的细胞凋亡,ATM没有影响p53激发的转录信号,但是它阻断了自噬活动,从而维护了线粒体的平衡并且抑制活性氧的水平。
总体来说,这些观察意味着p53媒介的生理反应不是一个简单的开和关。
p53显然深陷在一个由多种调节子和效应子构成的密集而且互相关联的网络中。
细胞的背景因素(细胞种类,表观生物学状态,组织微环境,激活信号)对p53活性的生化特性和p53反应的生物结果都有关键性的作用。
根据定义,肿瘤抑制基因调节那些限制不正常细胞增殖的生理过程,这些基因失活会促进肿瘤形成和生长。由于p53能够控制多种生理过程,哪些效应功能对抑制肿瘤来说最重要一直是一个争论不休的话题。细胞衰老和凋亡显然在肿瘤中存在而且当这些生理过程被激活时确实能够起到抑制肿瘤的作用。但是,近年来的研究表明抑制肿瘤并不一定需要细胞凋亡或者衰老。在有些情况下,p53媒介的非经典功能可能更加重要。
对于肿瘤抑制来说,唯一的检测指标是这个基因能够在体内削弱肿瘤的产生或发展。
从这个角度来说,p53基因敲除(p53 knockout)小鼠是个有力的模型,这些小鼠无一例外地会得上胸腺淋巴瘤(thymic lymphoma)。为了研究哪些p53媒介的功能对肿瘤抑制最为关键,各种不同的转基因小鼠模型被创造出来。如果消除p53控制的某些功能导致肿瘤的生成,那么被消除的过程就对p53抑制肿瘤的活性非常重要,反之则不重要。
一种研究方式是比较p53缺失小鼠和p53靶点基因缺失小鼠在肿瘤生成和病理方面的差别。
例如,p21、Puma或Noxa缺失小鼠并不会得上胸腺淋巴瘤,这意味着p53媒介的细胞周期停滞和细胞凋亡可能对肿瘤抑制来说不是必不可少的。但是p53的靶点基因通常在细胞中有一定基准水平的表达,例如p53缺失的小鼠中p21基因显然没有缺失。于是,
这种研究策略可能导致某些p53效应子对无效表型(null phenotype)的贡献被高估。
反过来说,由于多个效应子在媒介大多数p53的功能,缺失其中一个效应子的小鼠模型可能无法让p53的效应过程完全失活(例如p21缺失不能完全阻止p53介导的细胞周期停滞)。于是,
这种研究方式也可能低估某些p53靶向过程对肿瘤抑制的影响。
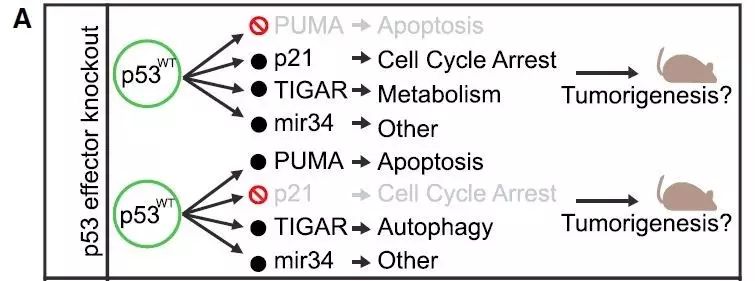
▲
肿瘤抑制机制研究:敲除p53靶基因
另一种分割p53功能的研究方法是,使用有选择性地保留或者丢失一部分p53靶点基因的能力和活性的突变模型。
例如,从肿瘤中获得的
p53R175P
和
p53E180R
等位基因会导致细胞凋亡的功能失常,但是保留了引发细胞周期停滞的能力,携带这些基因变异的小鼠与p53缺失小鼠相比无肿瘤生存期得到了延长。这意味着不同的p53突变体可能有选择性地影响了下游的效应信号通路。另外一种方法是用人工合成的结构功能突变体(structure-function mutant)来扰乱p53的转录蛋白域或者让乙酰化过程失常。这种方法至少可以在体外将p53的重要功能分割开。不过结构功能的研究策略也有它的缺陷。携带p53突变的蛋白与野生型相比可能更为稳定或更不稳定,因此导致的不同表型可能是由于蛋白数量上的差别,而不是蛋白功能上的差别。大多数结构功能突变只在少数几种细胞中被研究过,这些研究结果可能无法被推广到所有组织的肿瘤生成中。
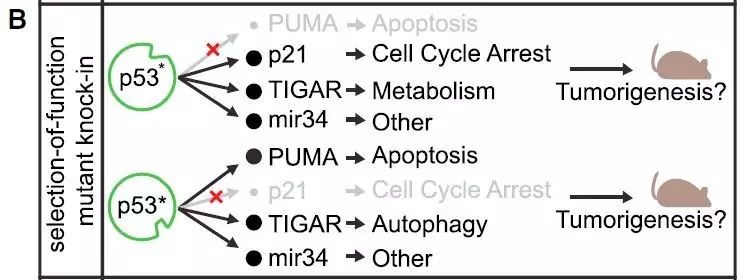
▲
肿瘤抑制机制研究:
突变p53靶基因
第三种研究方法是利用携带能够被重新激活的p53等位基因的小鼠肿瘤模型。
目前的研究结果表明,恢复p53功能可以产生强烈的抗肿瘤反应。例如在表达Myc的B细胞淋巴瘤模型中,恢复p53功能导致大量细胞凋亡。而在肝癌模型中,恢复p53导致细胞衰老。在其它情况下,重新激活p53可以引发细胞分化和自我更新的丧失。虽然在肿瘤模型中重新激活p53的后果并不见得等同于肿瘤发生时丢失的生理过程,这些研究重申了背景因素对p53功能反应的影响。
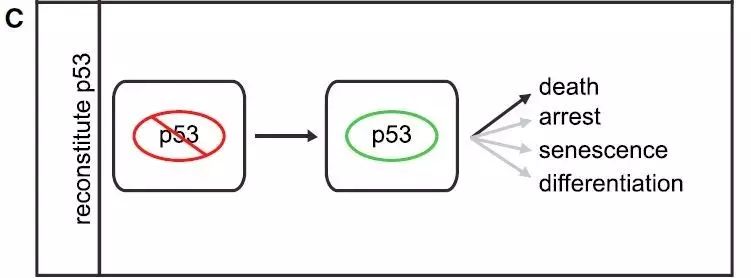
▲
肿瘤抑制机制研究:
恢复p53基因功能
那么,到底哪些p53活性对于肿瘤抑制来说是最重要的呢?显然,上述研究的缺陷说明我们不能忽视背景因素而进行概括。接受背景因素的重要性可以让我们发现针对不同肿瘤的抑制方式,并且可能导致未来根据不同肿瘤的特征来选择恢复最相关的p53功能。
TP53
基因位点在肿瘤中的变异方面的研究进展,不但没有简化我们对p53变异如何促进肿瘤生成的理解,反而让我们的理解更为复杂化。
TP53
基因变异中最常见的也是被研究得最为彻底的是位于DNA结合蛋白域的错义突变(missense mutation)。这意味着p53的这一特征对肿瘤抑制非常关键。目前的教条是把p53分为野生型或者突变型两大类,
但是
TP53
突变可以以许多形式出现,并且伴随着不同的协同突变和多种多样的等位基因组合模式。
因而可以导致非常有趣的功能性与表型分支。
基因组测序已经证实,
大约一半肿瘤至少携带一个p53变异
,
但是不同类型的肿瘤之间携带p53变异的频率和分布有非常大的差别。
大多数在TP53基因中出现的 SNVs是错义突变,其中25%属于5种热点突变。令人意外的是,大约25%的
TP53
突变是会导致编码截短蛋白的无义或移码突变,剩下的是剪接点SNVs和生理意义不明的片段插入或缺失。虽然有几种渠道可以让第二个
TP53
等位基因失活,但是这通常是由于片段删除(segmental deletion)而造成的。这些被删除的片段大小不一,出现的频率和SNVs出现的频率相当。大约只有25%的肿瘤携带着典型的p53错义突变/片段删除组合。
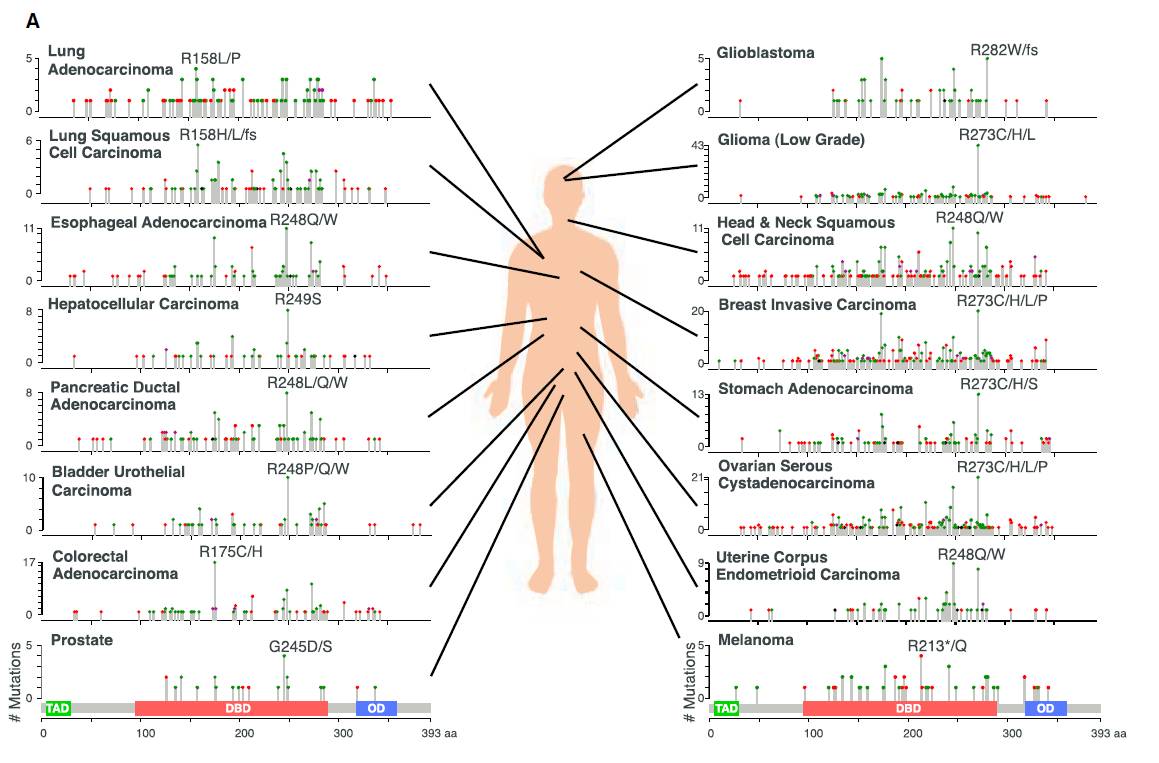
▲
16种癌症类型中的常见
TP53
突变
癌症基因组项目帮助我们进一步了解
TP53
变异图谱和它与其它体细胞基因变异之间的关系。在某些癌症中,
TP53
基因突变通常与激活
KRAS
的基因突变和MYC扩增同时发生。而且如前所述,
TP53
基因突变经常与高频率的基因拷贝数目变异联系在一起
,例如在卵巢癌和复杂核型的急性骨髓性白血病中这种情况经常发生。
对
TP53
在不同情况下的变异进行归类,可能让我们判断特定的变异是否反映出功能选择,或者只是肿瘤发生时存在的不同突变产生方式。
TP53
中特定的突变特征一部分是由于突变产生的来源造成的。
例如,在肝癌中常见的
R249S
突变是由于黄曲霉毒素导致的G变为T的转换,而在黑色素瘤中的
R123*
突变是由于紫外线导致的C变为T的转换。有些经常出现的C变为T的突变是由于胞苷脱氨酶的作用,它为抗体多样化提供内在的基因变异。
过去25年里的试验数据同时显示,
某些TP53突变为获得功能性突变(gain-of-function mutation)
。这类突变蛋白最显著的表型是它们能够增强肿瘤的侵袭和转移,有些时候它们会增强药物抗性,表观遗传重组,和血管增生。虽然这些获得功能性突变可以有多种活性,但是最主要的规律是这些突变会对抗野生型p53的功能,或者加重p53缺失造成的结果。
一个p53突变能够造成特殊的表型并不足以证明这个突变是获得功能性突变。理论上讲,携带p53突变的等位基因可以导致功能减弱,功能分离或者功能获得。减弱野生型的功能(下图A)可能基于对p53水平不同程度的抑制造成功能特异的表型(下图B)。例如,靶向p53的shRNAs可以将p53敲低到不同水平,这会对小鼠模型中p53功能和淋巴瘤的产生造成不同的影响。但是只有完全敲除p53可以导致染色体的不稳定。功能缺失是癌症相关的p53基因突变中常见的一个特点,表现在大多数突变无法引发细胞凋亡。功能分离性突变表现在突变的p53基因保留了一部分与下游靶点的相互作用,例如前文描述过的p53R175P突变(下图C)。最后的一种类型的获得功能性突变(下图D)在下文中将继续描述。
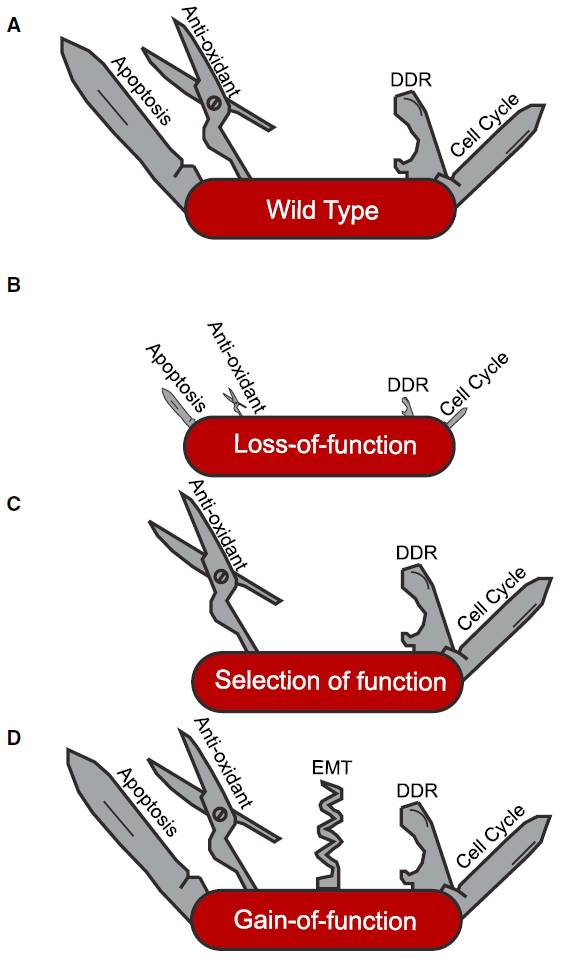
▲
不同机制可导致p53突变的不同表型:(A)野生型,(B)功能丧失或部分丧失,(C)功能选择,(D)新形态/功能增益。
p53突变引发致癌效果的机制还没有得到完全澄清
。第一,有些保留了残余激活转录功能的p53突变蛋白可能激活全新靶点。例如,p53突变型可以通过激活MLL1/2和MOZ的表达来影响染色质形态。第二,有些非结构化(unstructured)的p53能够隔离其它蛋白,并且在有些情况下让p53突变蛋白能够与p63或p73结合,导致受体酪氨酸激酶信号通路的变化,从而促进肿瘤侵袭和转移。最后,p53突变蛋白可以与SWI/SNF复合体合作来提高调控血管增生的VEGFR2的水平。
虽然通常的假设是
TP53
截短突变会造成无义等位基因,
但是最新研究表明这些突变可能造成新功能的产生。
比如,有些无义突变,特别是在6号外显子上的无义突变的频率比预想的要高。其中有些突变让截短的p53突变体能够与错义突变蛋白一样能够促进肿瘤侵袭和转移。
除了不同p53 SNVs造成的异质性以外,
人体17p染色体上不同程度的片段丢失也会造成p53基因剂量上不同程度的改变。
而且在肿瘤中观察到的人类17p染色体的片段丢失经常导致p53基因以外其它癌症抑制基因的缺失。模拟17p13的17p染色体片段丢失与p53缺失相比,在小鼠中造成侵袭性更强的肿瘤,原因是很多需要两个拷贝才能抑制癌症的基因丢失了一个拷贝。
这些观察结果表明与CNVs关联的独特生物学和进一步对其进行分析的重要性。
综上所述,我们对
TP53
变异复杂性的了解正在改变我们如何看待这一“在人类癌症中最常见事件”对肿瘤发生的影响。虽然不容质疑的是最显著的生物学后果是由于p53蛋白失活而造成的,但是很明显TP53基因变异和17p片段缺失对癌症表型的影响超过单单造成p53缺失。由于未来临床决定的产生将更依靠基因组数据,
现今将肿瘤简单分为“p53野生型”或“p53突变型”的分类方式必须改变
。
p53在肿瘤抑制方面的强烈功效和癌症中p53变异出现的高频率激发了许多针对p53信号网络的癌症疗法的研发。研究表明对传统化疗的有效反应基于野生型的p53。例如,治疗急性早幼粒细胞白血病(acute promyelocytic leukemia)的视黄酸与砷疗法的成功取决于p53媒介的细胞衰老。由于
TP53
基因突变会让野生型的p53蛋白失活,因此这些基因突变普遍被认为无法用药物靶向(undruggable)。然而,其它靶向p53突变蛋白,p53调控子,或者
TP53
基因突变在癌症中造成的薄弱环节的努力开始显现出成效。
目前利用我们对p53生物学理解进行抗癌药物研发中,进展最快的项目是
抑制携带野生型p53的肿瘤中的MDM2
。以Nutlin为首,一系列抑制MDM2和MDMX的小分子和肽基抑制剂被开发出来。它们通过靶向MDM2中与p53的结合位点来防止p53的降解。MDM2拮抗剂在白血病和脂肪肉瘤中已经完成一系列临床1期试验。虽然这些剂量递增试验不能对药物疗效作出结论,但是在大多p53野生型样本中观察到了p53靶点基因的表达量增加。而且在5~10%的患者中观察到了部分反应,这是一个好的预兆,因为这些患者中很多已经接受过多种疗法的治疗。
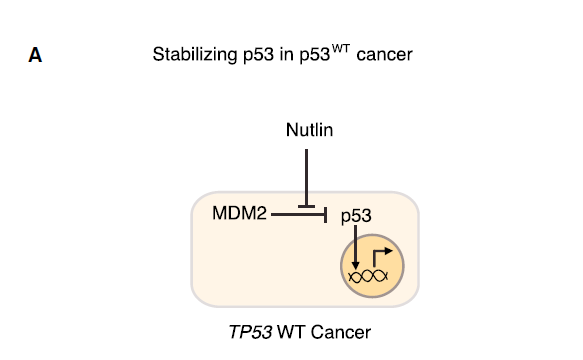
▲p53治疗策略:
在p53野生型肿瘤中保持p53稳定性
在另一种情况下,
MDM2抑制剂被用来减少化疗的毒副作用
。在这种策略中,这些药物被用来在正常细胞中提高p53的稳定性,从而触发暂时的细胞周期停滞。这一做法的目的是对于携带p53突变的肿瘤细胞来说这些药物对细胞周期周转没有作用。因为细胞毒性药物靶向积极分裂的细胞,这一策略可以提高化疗药物的使用剂量,在增强对分裂的癌细胞的疗效同时降低对细胞周期停滞的正常细胞的副作用。
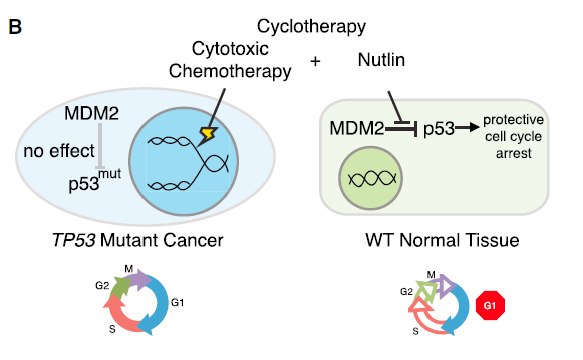
▲p53治疗策略:采用
MDM2抑制剂减少化疗的毒副作用
另一种吸引人的策略是
发现能够让p53突变蛋白重新获得野生型p53的肿瘤抑制活性的药物
。虽然达到这一目标的热力学要求是一个严峻的挑战,但是蛋白结构研究和电脑模型预测已经发现了多个策略在概念上证明这一方法的可行性。其中包括可以让非结构化突变蛋白结构稳定下来的小分子和多肽。一种名为APR-246的药物据称可以重新激活p53突变蛋白但是具有脱靶效应,该药物现在正在进行临床试验。其它直接稳定p53的DNA结合蛋白域的药物在临床前实验中表现出可喜的前景。被称为金属伴侣(metallochaperons)的药物能够帮助展开的蛋白(unfolded protein)与锌离子结合并且形成一个较为正常的构像,从而恢复与DNA结合的能力。而另一种策略利用了一个意想不到的发现,那就是有些p53突变蛋白有形成像淀粉样蛋白一样的聚合物的倾向,当聚合过程被扰乱后,p53的功能可以得到恢复并且在异种移植模型中触发肿瘤萎缩。
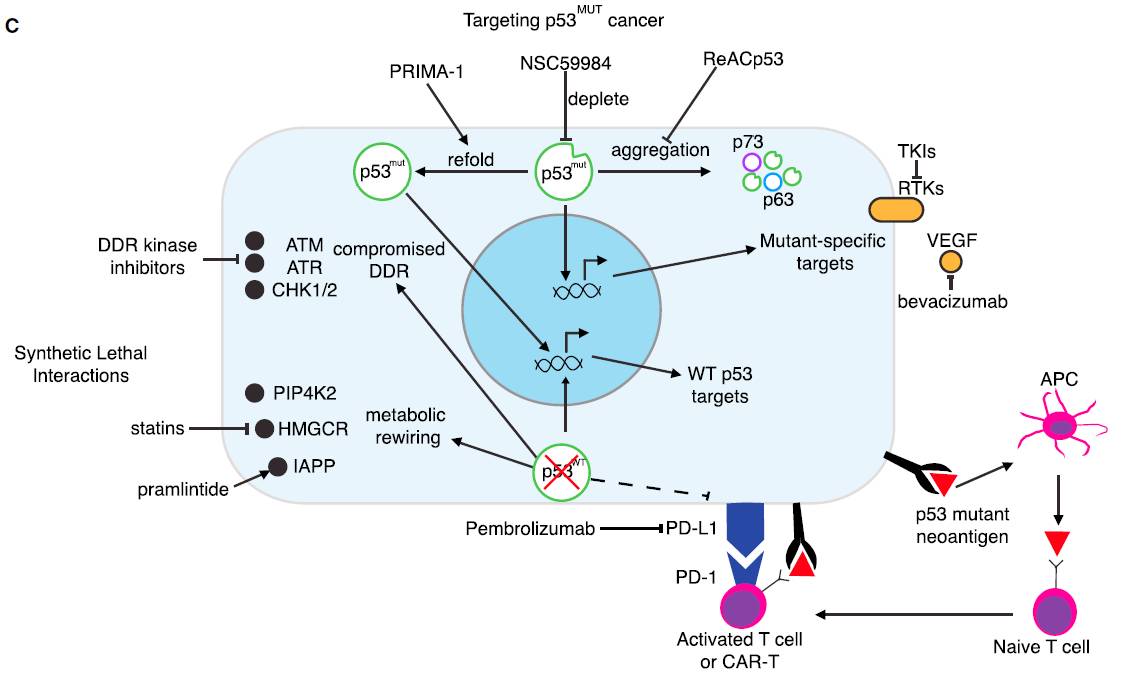
▲p53治疗策略:靶向p53突变的肿瘤
除了利用药物恢复突变蛋白中野生型的活性以外,
另外一种策略是抑制p53突变蛋白的活性
,因为有些肿瘤会依赖于p53突变蛋白的功能。在无法直接抑制p53突变蛋白活性的情况下,仍然可以有方法间接靶向p53突变蛋白促进肿瘤细胞侵袭、转移和存活的机制。
另一种直接攻击p53突变蛋白的方法是
利用它可能成为具有肿瘤特异性的新抗原(neoantigen)的潜力
。p53突变蛋白通常表达量很高而且具备抗原特性。而且针对p53突变蛋白的疫苗在小鼠模型中已经取得了一定成功。基于这个想法,多肽疫苗,病毒载体和树突状细胞疫苗已经进入临床1/2期试验。不管基于哪种平台,免疫疗法已经能够引发针对p53的免疫反应。
理论上逃脱免疫编辑的肿瘤更可能携带激发免疫反应的新抗原。因此将p53免疫疗法与增强T细胞反应的免疫检查点抑制剂结合起来,可能将针对p53的T细胞转化为抗癌武器。
另一个靶向携带p53突变蛋白肿瘤的具有吸引力的策略是
利用合成致死概念
。很多p53突变蛋白造成的弱点与DDR和新陈代谢相关。虽然p53缺失的细胞在面对损伤DNA药物时能够避免细胞凋亡,但是进一步破坏DDR系统会让携带p53突变的肿瘤对遗传毒性损伤非常敏感。因此,人们正在开发将造成DNA损伤的药物与抑制DDR系统中的ATM, CHK2, ATR和CHK1的抑制剂联合使用的策略。虽然加剧基因组不稳定性可能达到治疗效果,但是一个顾虑是削弱DDR导致的基因突变可能进一步加快肿瘤进化。
通过恢复野生型功能,抑制突变蛋白功能,或者靶向失控的免疫系统,有多种渠道可以靶向癌症中的p53信号网络。
目前使用这些策略时面对的挑战表明,进一步理解p53的基本生物学机制对未来临床应用的成功非常重要。
结束语
p53是一个让癌症生物学家着迷的蛋白。
对它的详尽研究增进了我们对基因调控机制和大自然防御癌症手段的理解。虽然对p53的海量研究有时自相矛盾,但是现在非常清楚的是细胞对p53激活的反应包含着触发因子,细胞谱系和细胞状态之间的复杂相互作用。这种依赖背景因素的p53作用方式虽然让寻找通用的p53媒介的肿瘤抑制机制困难重重,
但是它给予我们在癌症细胞中利用信号网络的机遇,让我们避免在所有组织中操作p53带来的有害影响。
点击
“阅读原文”
,即可访问原始论文页面。
参考资料:
[1] Putting p53 in Context







