在
上一篇教程
的最后,我写道「我们能够更加直观的观察到2个peak,基本确定2个多倍化事件」。这里就引出了一个问题,为什么我们可以根据peak来推断多倍化时间?在Lynch和Conery在2000年发表在Science的论文中,他们证明了小规模基因复制的Ks分布是L型,而在L型分布背景上叠加的峰则是来自于演化历史中某个突然的大规模复制事件。例如下图a中,实线是小规模复制的L型分布(呈指数分布, exponential distribution), 最初的峰可能是近期的串联复制引起,随着时间推移基因丢失,形成一个向下的坡。另一条虚线中的峰(呈正态分布, normal distribution)则是由全基因组复制引起。而b-h是一些物种的ks分布。
❝
注,Lynch和Conery这两人是最早一批研究真核基因组的复制基因的总体保留和丢失情况的研究者。
❞
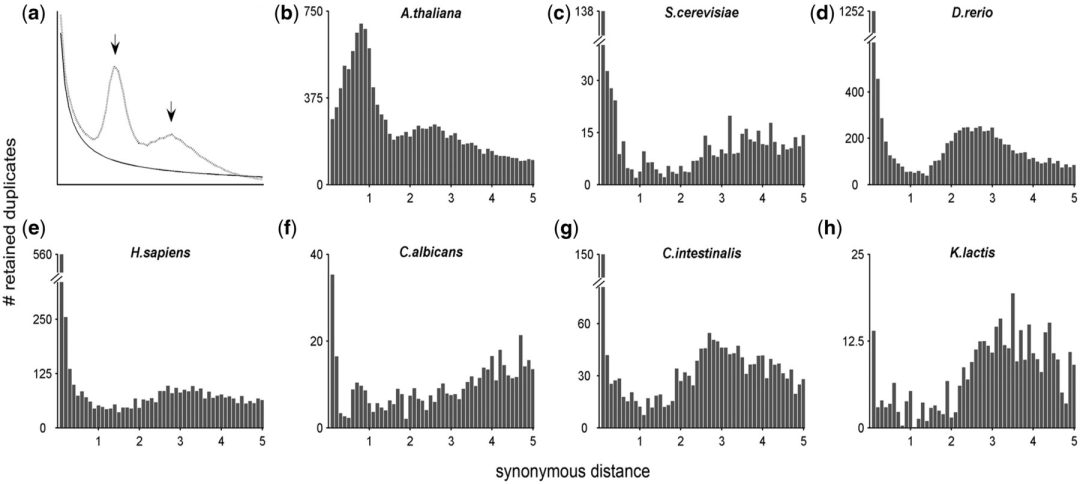 Kevin Vanneste et al. 2012 Molecular Biology and Evolution
Kevin Vanneste et al. 2012 Molecular Biology and Evolution
也就是说,我们可以通过对Ks频率分布图的观察来判断物种在历史上发生的基因组复制事件。同时又因为WGD在Ks频率分布图中表现为正态分布,那么我们可以通过对峰进行拟合来得到WGD事件所对应的Ks值。
使用WGDI对峰进行拟合需要注意的是,
「它一次只能拟合一个峰」
,因此我们需要先通过之前Ks可视化中所用到kspeaks模块(-kp)来过滤,接着用
「PeaksFit」
(-pf)模块对峰进行拟合得到模型参数,最后用KsFigures(-kf)将拟合结果绘制到一张图上。
过滤的目的是筛选出同一个WGD事件所形成的共线性区块,这可以通过设置kspeaks模块中和过滤有关的参数来实现
-
-
-
block_length: 共线性区块的基因对的数目
-
ks_area: 根据 ks_median 筛选给定区间的共线性区块
-
multiple: 选择homo的哪列作为筛选标准
-
homo: 根据homo,筛选给定区间内的共线性区块对
其中比较关键的是ks_area 和homo,mutiple, 前者确定ks的大致区间,后者则是更精细地筛选共线性区块。block_length可以过滤掉一些比较小的共线性区块,毕竟基因数越多,就越不可能是随机事件所产生的共线性。
通过观察之前Ks频率分布图,我们大致确定两个峰所对应的区间分别是0~1.25,1.25~2.5。
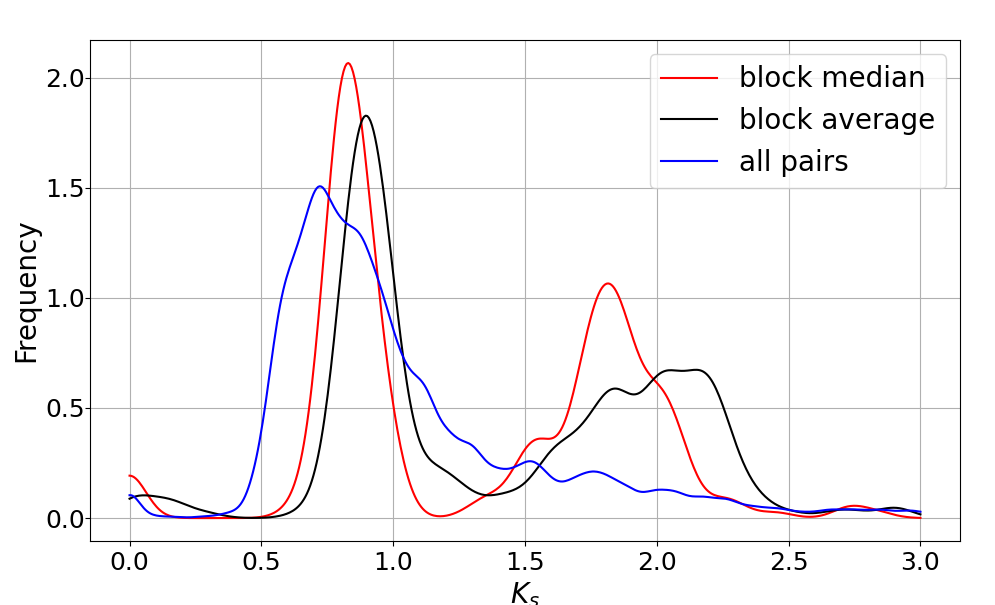 figure 1
figure 1
我们分别用
wgdi -kp \? > peak1.conf
和
wgdi -kp \? > peak2.conf
创建两个文件,主要修改其中的ks_area, homo都设置为-1,1 表示不筛选
# peak1.conf
[kspeaks]
blockinfo = ath_block_information.csv
pvalue = 0.05
tandem = true
block_length = 5
ks_area = 0,1.25
multiple = 1
homo = -1,1
fontsize = 9
area = 0,2
figsize = 10,6.18
savefig = ath.ks_peak1.distri.pdf
savefile = ath.ks_peak1.distri.csv
# peak2
[kspeaks]
blockinfo = ath_block_information.csv
pvalue = 0.05
tandem = true
block_length = 5
ks_area = 1.25,2.5
multiple = 1
homo = -1,1
fontsize = 9
area = 0,2
figsize = 10,6.18
savefig = ath.ks_peak2.distri.pdf
savefile = ath.ks_peak2.distri.csv
运行
wgdi -kp peak1.conf
和
wgdi -kp peak.conf
输出结果。下图中左是peak1, 右是peak2.
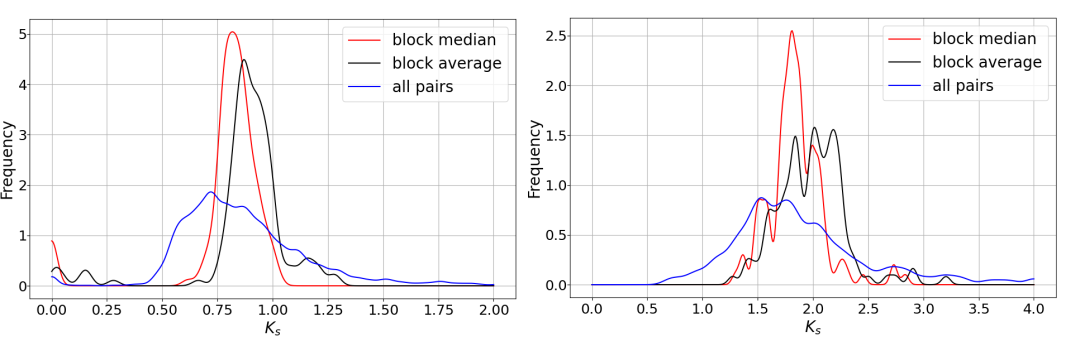 figure 2
figure 2
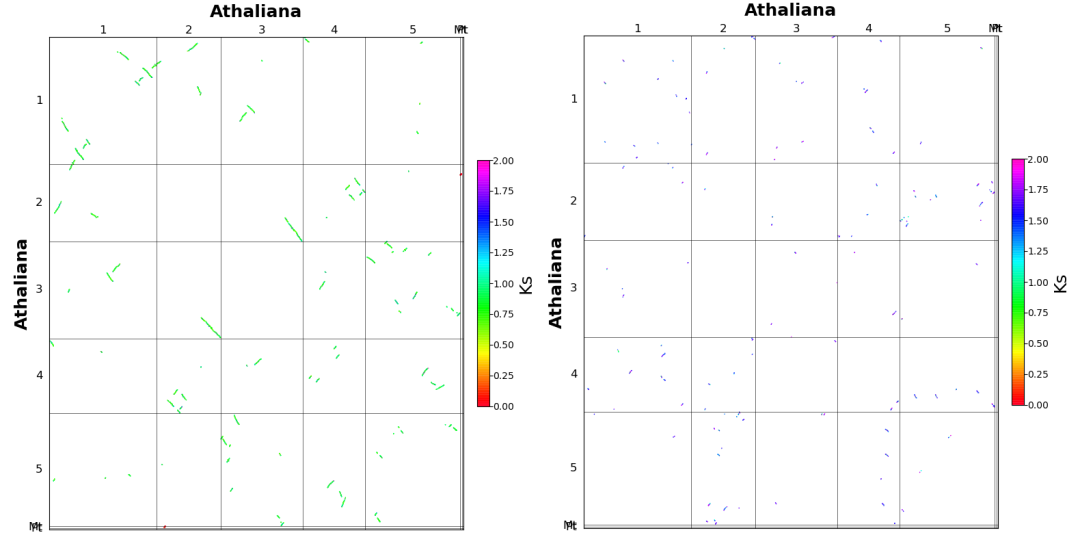
得到这个图之后,我们还可以进一步绘制Ks点阵图, 使用
wgdi -bk >>peak1.conf
和
wgdi -bk >> peak2.conf
增加配置信息,然后对其进行修改
# peak1
[blockks]
lens1 = ath.len
lens2 = ath.len
genome1_name = Athaliana
genome2_name = Athaliana
blockinfo = ath.ks_peak1.distri.csv
pvalue = 0.05
tandem = true
tandem_length = 200
markersize = 1
area = 0,2
block_length = 5
figsize = 8,8
savefig = ath.ks_peak1_dotplot.pdf
# peak2
[blockks]
lens1 = ath.len
lens2 = ath.len
genome1_name = Athaliana
genome2_name = Athaliana
blockinfo = ath.ks_peak2.distri.csv
pvalue = 0.05
tandem = true
tandem_length = 200
markersize = 1
area = 0,2
block_length = 5
figsize = 8,8
savefig = ath.ks_peak2_dotplot.pdf
运行
wgdi -bk peak1.conf
,和
wgdi -bk peak2.conf
输出结果。下图中,左边是peak1,右边是peak2.