在两年前,生信媛推送李广伟师兄写的binmap系列教程,尽管当时文章是我编辑的,但是其实对binmap并没有太多理解,直到最近做了几个相关项目之后,才感觉自己理解了binmap。这次重温的时候,我顺手将本文的代码整理成一个R包,明天将会使用我写的R包重现本文的结果。
数据的百度盘地址为: https://pan.baidu.com/s/1c2OvL3E 密码为
fm5l
百度链接这两年来一直都能用,从没有变过,反倒是我自己提供的GitHub地址失效了。
基因组时代,BSA,Binmap,GWAS是遗传学的三大宝刀。
-
BSA最省时、省力、省钱,但是需要你有
好的材料
,进行
合理实验设计
。很多同学都被测序公司骗,以为什么都能BSA,甚至自己都不清楚自己的群体分离比究竟咋回事,就混池测序,只会越混越乱。
-
Binmap是最
中规中矩
的实验设计,需要你构建群体,对群体中每个单株进行测序和考察表型,这有点小烦人。但是随着测序成本的降低,相对于BSA也多花不了几个钱,个人还是非常推崇这种稳扎稳打,步步为营的遗传学方法。
-
GWAS其实是最懒的实验设计(我指的是对某些异想天开的实验室)。为什么说他懒,因为很多人都是从别人那里拿来种质资源,考察表型,然后交给公司测序,拿来SNP数据,甚至后续分析都是公司搞定。很多都是发文章
一锤子买卖
,很难有下文,图的就是短平快。非常符合当下对SCI,IF充满着饥渴的科研环境。很多需要对材料群体结构进行精细考量,对关注的表型进行系统设计的环节被完全省略,完全变成了#比钱多,比材料多的比大小游戏。
小结
:相对简单粗暴的GWAS而言,BSA和Binmap都更需要关注一些的生物学问题,脑袋非常清楚的去发现,甚至创造材料,进行实验设计。要不很难有好的实验结果。当然我们也需要在群体水平和应用方向上挖掘信息,提高我们研究的意义和质量。
前段时间介绍过
BSA
定位过基因,今天就来讲Binmap。前部分是基本概念介绍,后面是代码实现。老少皆宜,适合睡前十分钟看完,然后动动手指头转发、点赞、打赏、拍砖等一系列睡前非理智行为。留在白天,在R中复制粘贴代码,十分钟之后,困扰你几年已久的binmap构建完成,几秒之后QTL扫描完成。
bin在基因组学上其实有两个概念,一个等同于大家熟悉的window(plink的官方文档有涉及,不知所云代表你不需要知道);另外一个侧重于遗传学,它是指在一个分离群体中,
没有发生重组的染色体区块
。后面一个概念有点绕。什么意思?
我们都知道一个个体要去生孩子,就要先发生减数分裂产生配子,减数分裂过程染色体会随机重组,发生交换,这个交换位点在染色体上是“随机的”。一个F1自交产生了200个小娃娃,这200个小娃娃的400条染色体就会发生很多次交换,但是注意这些交换位点是随机,那么就会有些区段,一次交换也没有,一次交换也没有,一次交换也没有。一次交换也没有怎么办?那就不换呗。不换怎么办?那这段就是一个bin呗。
沃特,还不明白?或者你本来明白,被绕的晕乎乎的。这个时候你室友叫你来斗地主,正和你意!第一盘,你上来就是双王,王炸下去,赢了一盘。然后第二盘,你叫了地主,翻起底盘,亮瞎了你的“人”眼,底盘留着双王。你室友肯定大叫,“这怎么洗的牌!牌没洗净,重来!” “啥,牌没洗净?!” 对,就是没洗净,上次连着,洗牌后,还连着,中间一点没交换交换。对!这就是bin啊,中间没交换。你恍然大迷瞪。哦,bin就是基因组上这一块,洗牌时候,没交换,没洗净,以至于出现在下一代时候,还连在一块没打断啊!
这个时候,你可能理解了概念,那咱就来一个专业点的小故事再来两个疗程,巩固巩固,看下图。
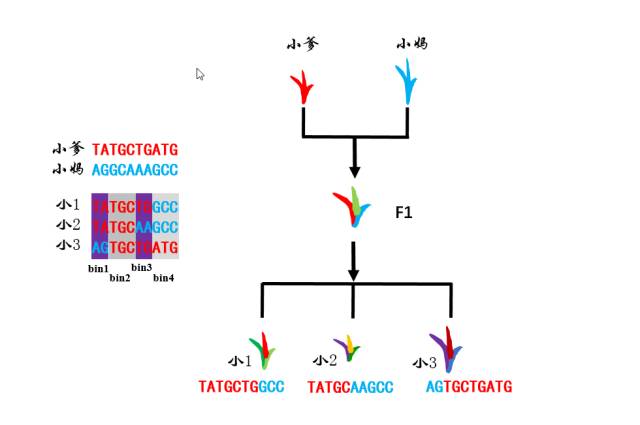
图1
:一个简单的F2群体信息
如图1“小”的单倍体小物种,染色体非常小,whole genome只有10个碱基。小爹,小妈的10个碱基都有差异。小爹小妈有一个孩子,这个孩子长大后,自交生下了3个孩子。这三个孩子就是一个小小的F2群体。三个孩子的染色体各发生一次交换,小1断点发生在第6、7个碱基之间,小2断点发生在第5、6个碱基之间,小3断点发生在第2、3碱基之间。通过比对之后,发现三个个体组成的群体中第1、2碱基之间没有发生交换,于是这两个碱基就是一个bin,第3-5个碱基也是一个bin,第6-7个碱基是一个bin,第8-10个碱基是一个bin。
如果我们把小爹基因型
code 1
,小妈基因型c
ode成 0
。三个个体基因型整理成如下格式:
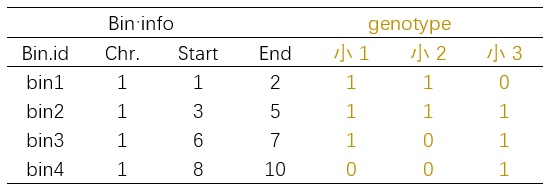
表1
:“小”物种三个F2个体的binmap
binmap构建的流程:
-
选定一个单株,通过plot查看chr01上SNP情况,了解大致的纯和区域和杂合区域。
-
通15个SNP一个window的滑动窗口,从头到尾滑动,通过15个SNP sum值得到这个材料chr01上转换成window的基因型
-
通过window周围的基因型信息,对错误的基因型进行修正
-
根据window基因型信息,判断染色体上的重组断点,即构建选定单株的chr01上的bin
-
对群体中的每个材料chr01进行bin推定,然后把群体中的这些断点,沿着chr01从头到尾排列,整合成出来chr01的binmap(回看图1)

下面是一个实际的案例,数据来自水稻的一个F2群体,一共167个个体,表型是株高,通过RADseq获得了每个个体的SNP数据.
数据链接:百度云盘(
阅读原文
) 密码:fm5l。当然,我们还把数据上传到GitHub上,可以在R里面获取数据。
为便于大家学习,对数据进行了初步整理:每个SNP根据亲本基因型,P1基因型code成0,P2基因型code成2,杂合基因型code成1,NA为缺失。部分数据图示如下
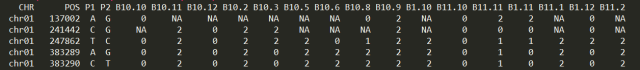
表2:水稻167个个体的F2群体chr01SNP部分数据
这里仅用第一染色体的9875个SNP来学习, 详见文件。这种格式怎么整理?其实这是数据的基本处理,exel可以搞定,请自学两天的R,或者请多来生信媛学习,另外测序公司可以帮你搞定,搞不定果断怼他。
下面开始构建binmap,我们是逐个材料,逐条染色体开始构建。让我们先开始分析B10.10。
数据分为两类:每个样本表型数据以及每个样本的SNP数据(约等于基因型数据),遗传定位的目的主要就是将表型于基因型一一对应。
library(readr)
library(tidyr)
library(dplyr)
snp_url 'https://raw.githubusercontent.com/xuzhougeng/zgtoolkits/master/chr01.snp.csv'
snp_data snp_df
phe_url 'https://raw.githubusercontent.com/xuzhougeng/zgtoolkits/master/phe.csv'
phe_data
简单的两行代码(加载包不算),这里是
重点
,敲黑板,睡觉的同学醒醒,我们需要盯着画出来的图2看上两分钟。
library(ggplot2)
snp_chr1_B10.10 p1 p1 + scale_x_continuous(labels = function(x) return(paste0(x/1000))) +
xlab("position") + ylab("SNP type")
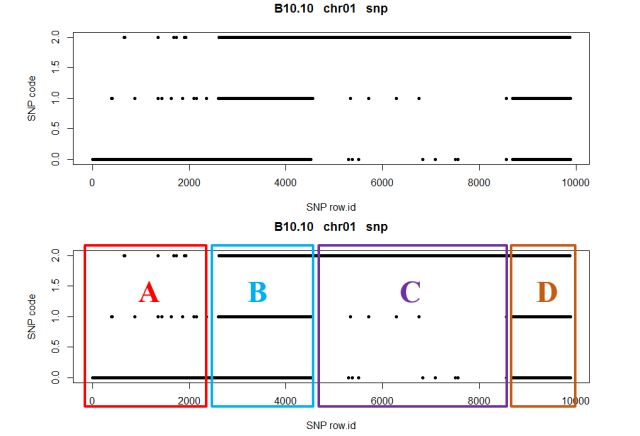
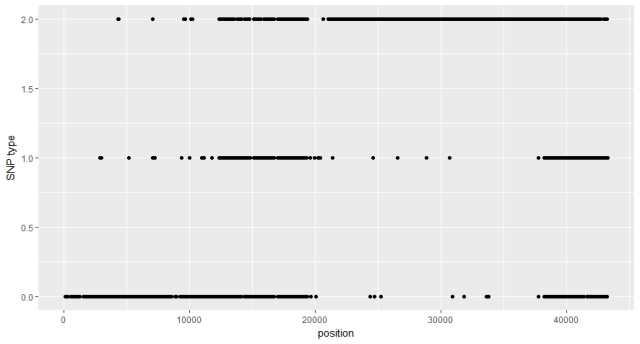
图2:B10.10个体,chr01 SNP情况
对于B10.10这个个体,chr01基本分为四个区域,分别为ABCD:A区域,大部分是亲本1的基因型,即SNP状态是0;区域B,SNP0,1,2状态都有,也就是两个亲本和杂合都有,区域C主要是亲本2基因型;区域D和B类似。
为什么会这样呢?我们一般对于一个群体的每个个体是
低覆盖度测序
的,注意“
低覆盖度
”.
那么对于
A区域
可以理解为,这一块就是亲本1的基因型,至于散落的code的1和2的状态,主要是
测序错误
和
重复区域的snp calling的错误
导致。
对于
B区域
,这是一个杂合区段,如果两条染色体上我们都测到了,那么这个SNP就会检测出来是杂合的,但是如果只测了一倍的覆盖度,也就是只测到了一条染色体,就测成纯和基因型了,就是随机测成亲本1或者亲本2,所以才会出现三种情况都有。对于区域C,这一块就是亲本2的基因型,区域D也是杂合区域,同样有些错误的基因型。
以上问题主要是因为我们测序的低覆盖度造成的,我们通过提高覆盖度可以解决以上问题,但是对于我们F2群体的使用目的也就是
初步定位
,没必要花几百万干这种事,我们可以通过另外一种方法来矫正这种错误,那就是使用window。
也许只接触一天,你不知道一个人是究竟坏人还是朋友,但是接触上十年了,基本上能让一个人看透了吧。
正所谓,日久见人心,
SNP多了不怕错
。我们我们用一个window囊括15个SNP,25个SNP…..,我们来看这个window的基因型,而不是每个SNP的基因型,这样即便错也错不到哪里去。
我们的基因型已经code成数字形式,那么我们就设一个window15个SNP(跳过缺失的SNP),Sum一下,那么这个window的值应该是在0-30之间
-
等于0,即P1的基因型;
-
等于30,即P2的基因型;
-
杂合基因型应该在15左右波动。
第三步:构建Windows基因型
把SNP基因型转换成window基因型,15SNP/per window
wind_sum % mutate(group = as.numeric(rownames(.)) %/% 15 + 1) %>%
group_by(group) %>%
summarise(start = min(POS), end=max(POS), code_sum = sum (code)) %>%
ungroup()
ggplot(wind_sum, aes(x=start, y = code_sum)) + geom_point() +
xlab("Windows Id") + ylab("SNP code sum") +
scale_x_continuous(labels = function(x) return(x / 1000))
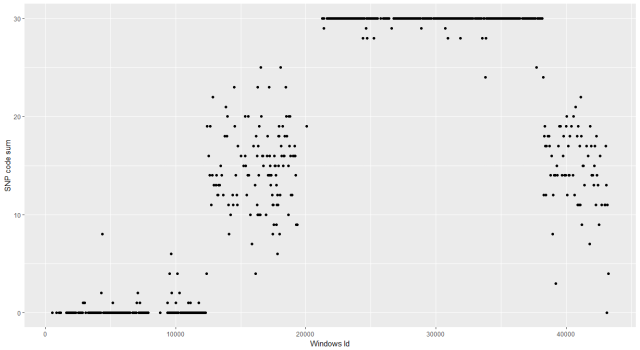
图3:每个window的SNP sum值
由图3可以看到,图2经过window处理之后,杂合区域不再是三条线,而是围绕15上下波动的一些点。亲本1纯和区域和亲本2纯和区域,也都是预期中的在0和30处的一条直线,外加偏离的一些点。虽然我们期望亲本1基因型值为0,但是总会有个别一些window,会偏离出来,对于亲本2也是这样。
于是需要我们进行技术处理一下,把Sum值小于等于6的window,都当成是亲本1基因型,code成0;把大于等于24的,都当成是亲本2基因型,code成2;把6~24之间的code成1,即杂合区段,我们来看看效果。
wind_geno ifelse(code_sum < 6, 0, ifelse(code_sum > 24, 2, 1 )))
ggplot(wind_geno, aes(x=start, y = code)) + geom_point() +
xlab("Windows Id") + ylab("SNP code sum") +
scale_x_continuous(labels = function(x) return(x / 1000))
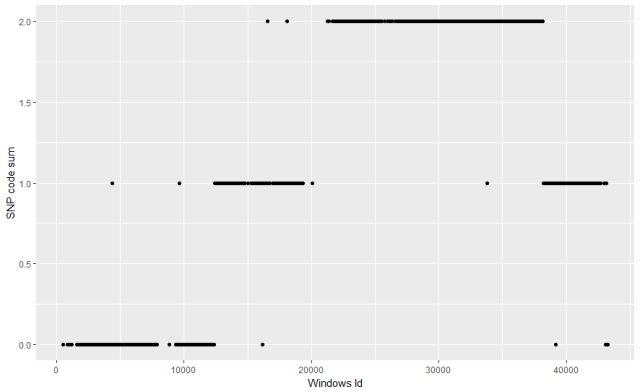
图4:每个window的基因型plot
由图4可知,图3的杂合区域,基本转成杂合基因型1,但是亲本基因型区域和杂合区域,总有一些点不合群,杂合区域离群点尤其多,这也跟杂合区域call基因型准确率更低有关。那么我就需要对这些潜在的错误window基因型进行修正,修正的方法也很简单,用它周围的window的基因型替换即可(上游下游问题都不大)。
fixgeno.func function(w.geno,fix.size=NULL){
wind.geno.rle error.id for(i in error.id){
left.id 1:i]) - wind.geno.rle$lengths[i]
right.id 1:i])if(i==1){ w.geno[(left.id+1):right.id] 1]
}else{
w.geno[(left.id+1):right.id] }
return(w.geno)
}
wind_geno$fix 10)
ggplot(wind_geno, aes(x=start, y = fix)) + geom_point() +
xlab("Windows Id") + ylab("SNP code sum") +
scale_x_continuous(labels = function(x) return(x / 1000))
上面代码用到了一个关键函数rle,例子一下
rle(c(1,1,1,2,3,3,3,1,1))
#Run Length Encoding
# lengths: int [1:4] 3 1 3 2
# values : num [1:4] 1 2 3 1
它返回一个向量某个元素重复的次数。基因型值应该是一连串的0,1,2值,偶尔有一些错误的乱入。那我们fix前的基因型看一下
rle(wind_geno$code)
#Run Length Encoding
# lengths: int [1:21] 42 1 64 1 54 45 1 10 1 37 ...
# values : num [1:21] 0 1 0 1 0 1 0 1 2 1 ...
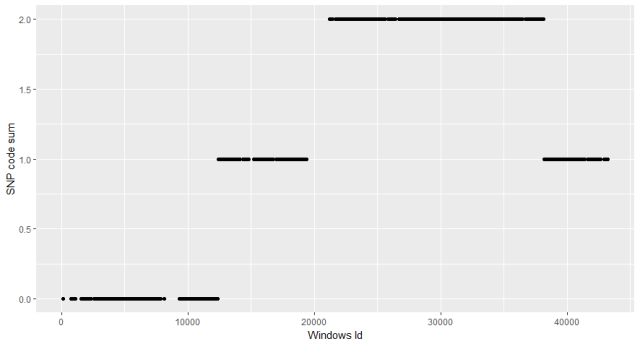
根据结果我们知道,在连续出现42个0之后,出现了1个1,然后又是连续64个0。也就是图4中,亲本1基因型区域中第一个乱入的杂合点(其实是6个点画到一块了)。我们就对这个错误的基因型进行替换,在这里我们写了一个fix的函数,可以选取从小到大的fix size从小到大,逐渐修正错误的基因,逐步打磨,得到比较靠谱的基因型结果, 如图5.
如上面结果所示,我们已经得到了B10.10这个材料第一条染色体的bin情况,那么我们一次在R里写一个循环可以把剩下的166个材料,剩下的其他染色体bin情况分析一遍,然后信息整合,就得到我们想要的binmap。
现在把我们从头开始的信息在一起回顾下:
bin_map 'method', value='value', code_sum:fix)
p1 "free")
print(p1)
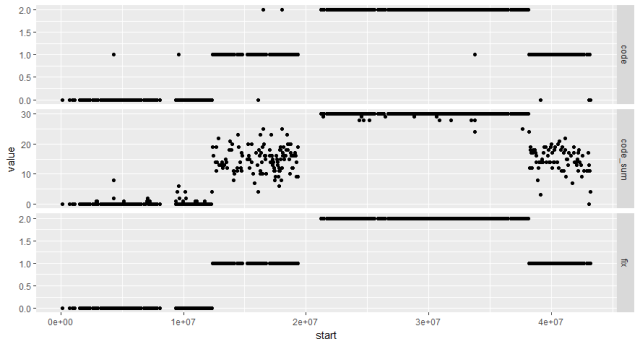
下面我们就是用一个向量,记录167个材料,chr01上所有的断点,每两个断点之间就是一个bin(回顾图1),整合信息,就得到了一个chr01的物理binmap。
运行下面代码,去泡杯咖啡,不出意外的话,回来之后167个材料,chr01的binmap(物理图谱),和一个pdf文档已经躺在你的文件夹里了:
setwd("F:/Data/binmap学习/")
source("binmap构建.txt")
那么构建完之后,我们的binmap质量怎么样的,那就把表型导进来扫个QTL试试吧。
phe_url 'https://raw.githubusercontent.com/xuzhougeng/zgtoolkits/master/phe.csv'phe_data
p 4:170],1,function(x){
lm p.t 5][1]return(p.t)
})
par(mfrow=c(1,1))
plot(-log10(p)~c(1:length(p)),pch=20,xlab="Chr01 bin id",main="chr01 株高QTL ANOVA 扫描结果",type="l",lwd=2)
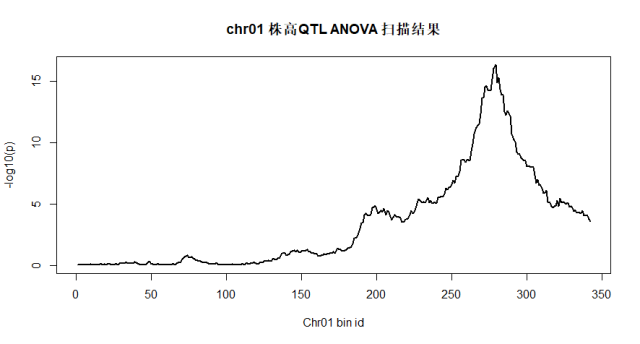
碰巧的是水稻里的绿色革命基因,sd1 LOC_Os01g66100 物理区间是 38382382-38385504,距QTL最显著位置只有258kb的距离,对于一个只有172个个体的F2群体而言,结果是不是已经很不错了。
bin map 教程都出来了,快转发这篇文章吧!

编辑:徐洲更





