作者:仲夏秋夜云 来源:蒲公英
访问对象:谢洋博士

对象简介:谢洋博士,QuintilesIMS中国区卫生经济与真实世界证据总监,负责中国地区的卫生经济学与医疗大数据研究工作。2017年加入QuintilesIMS。加盟QuintilesIMS之前,谢洋博士曾历任Pharmerit中国区总经理,默沙东总部亚太与新兴市场卫生经济与真实世界证据总监。谢洋博士还曾于美国爱荷华大学(University of Iowa) 担任卫生经济学与卫生政策助理教授,从事卫生经济学、卫生政策以及医疗大数据相关的科研教学工作。共发表过20余篇SCI国际期刊论文,近30个国际学术会议报告,并曾多次受邀于密歇根大学、中国药科大学等高校开展讲座。谢洋博士于纽约州立大学石溪分校获得经济学博士及公共卫生硕士学位。
1、任何建立标准模型的科学研究都不可能百分百反应事实真相,那么有没有数据显示III期临床试验和真实世界数据差异有多少?
正是因为没有一种研究方式可以同时完美地既满足对比性临床试验的内部有效性(internal validity)要求,又满足反应更广泛人群在日常生活中的实践的外部有效性(external validity)要求,现在大家才越来越认识到以真实世界数据(Real-World Data)和基于RWD产生的真实世界证据(Real-World Evidence)对临床试验数据进行补充的重要性。
真实世界数据和临床试验的核心差异来自于数据获得的场景不同。我们知道III期临床试验的目的是进一步在扩大的人群范围中评估药物的安全性和有效性,参与的患者都是经过严格的纳入排除标准选定的,且试验过程中对于药物用法用量,依从性等都有既定的规范或限制,尽量避免了个体差异对评估药物试验结果的影响。
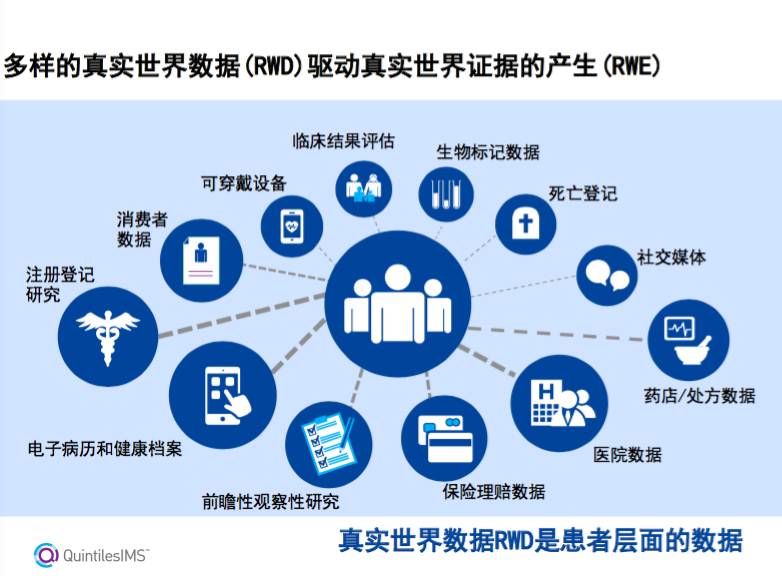
而对于真实世界研究而言,研究的实施场景为真实的临床实践环境,研究对象的选择通常不加特别的限制条件。可以说,III期临床试验中得到的数据和RWE是从不同的角度和出发点来反应药物作用和效果,并对药物使用或是临床实践的不同方面进行指导,以及对于III期临床试验的补充。
举个简单的例子,一些需要数年观测才能获得足够统计意义的发生率较小的不良反应信息很难通过3、5年,几百上千病人的RCT得到,而真实世界数据则可以让我们在几十万甚至上百万病例的多年观察中获得需要的信息,类似这样的情况正是FDA在去年《新英格兰医学杂志》上发表的《真实世界证据——它是什么以及它能告诉我们什么》一文中所强调的RWD的价值所在。
2、有学者认为III期临床研究无法反应真正的用药环境和人体差异,所以应该在II期过后,有一定的安全性评估保障后,直接投放市场,直接用RWE取代III期临床,您怎么看?
首先,III期临床试验是在评估药物安全性和有效性过程中非常关键的一个环节。对于在什么样的情况下,一个新药可能在II期过后先上市,用RWE在一定程度上取代III期临床试验,主流的学术讨论是做了严格界定的。如果盲目的直接在II期临床试验后将药物投放市场,带来的负面影响和风险是非常严重的。另一方面,对于某些特殊的药品,如缺乏有效治疗手段的罕见病的孤儿药,临床试验过于困难或耗时太长,或者是漫长的临床试验将导致一些有迫切医疗需求的病人无法得到治疗等,因此,很难完全按照评审要求搜集足够数量的患者进行大规模的III期临床试验,在这种情况下,通过RWD对临床试验进行补充而加快特定药物审批的探索,则是很有意义的。
欧洲药品管理局EMA在2014年EMA提出了企业药品评审过程中的“Adaptive Pathway”(可译为自适应路径),并在2014至2016年间展开了探索性的研究。具体来说,Adaptive Pathway指的是在药品评审过程中采用分步骤、循环对方式进行。即根据RCT的结果对药品进行初评,如果初评通过则允许在小范围、特定的患者群中使用,再根据真实世界实践中得到的证据对初评结果进行修正。目前EMA为期两年的探索性项目已经结束,EMA也根据项目的结果对RWD的使用做出了一定的总结,包括RWE的作用,允许使用RWE补充临床试验证据的条件等。
美国2016年底颁布的《21世纪治愈法案》要求FDA在未来的2-5年内出台关于在RWE在药物评审中使用的方案,且说明了RWE在药物评审中的两个用途:一是支持已经获批上市的药物扩大其适应症范围;二是支持已获批的临床试验的相关需求。这一法案的出台也意味着美国首次明确了RWE在药物评审过程中的作用。
现实当中,学术界的探讨尺度可能会比监管机构的探索要更大胆。如何平衡某些病人群对于新的治疗的需求和对于安全性风险的控制,以及如何控制药物研发时间与成本(因为最后这些成本的增加还是会以更高价格的形式由病人来买单),这是值得所有利益相关方一起探讨的。
3、RWE目前的研究数据来源是什么呢?和我们目前理解的IIIb段和IV期数据收集有何差异?
RWE的数据来源非常广泛,既可以是基于研究产生的数据,也可以看来自非研究信息。具体来说包括了:电子病历、医保数据库、病例报告、公共卫生调查、公共健康监测、电子设备和app、患者登记项目,甚至社交媒体等,当然在正常医疗环境下的IIIb或IV期的数据(相对于严格控制的II期、III期RCT)也属于真实世界数据的范畴。
在对RWE数据的收集和利用方面,更需各个相关方从药物开发生产到管理整个流程中通力合作。通过这种与价值链上各个有关方的合作,能够帮助企业了解药物真正的市场需求、治疗方法的选择、临床试验方案的制定等,加速审批和上市的进程。
4、RWE对于临床试验的实施,有怎样的优化指导作用?
RWE是真实世界环境下产生的证据,对象是更广泛的患者人群而非特定的患者,也并未受到过多的人为干预。 从真实世界数据中得到的结论可以对药物有效性、安全性进行补充,反映广泛的异质性群体的真实治疗情况,指导药品在不同人群中的使用效果,促进转化研究,发现最优效果人群、剂量建议等。目前阶段,以下两类是在利用RWE优化临床试验方面,QuitintilesIMS可提供的已较为成熟的业务:
减少临床试验设计缺陷: 如通过连接电子病历和生物标志物数据库, 来扩大II期临床试验的对象人数,提高试验结果的解释能力及对后期临床试验设计的指导; 或是通过RWE建立模型来优化试验设计(超过30%的临床试验是由于设计缺陷而非后期操作等原因失败的);
优化临床试验患者招募流程:III期临床试验中,患者招募往往是一个比较困难的问题。通过RWE了解目标患者分布,加速患者招募过程。通过挖掘真实世界数据,可以提高患者招募效率(enrollment rates)而这一过程通常的花费通常占整个临床试验的30%左右, 通过整合电子病历(EMR)和电子病例报告表(eCRF),可以节约约30%的成本。
除此之外,RWE的用处还包括了对上市后药品和器械的评价、临床试验指南的制定与修正、卫生技术评估、医保决策等。

5、目前国内RWE研究和欧美的研究环境差异在哪里?瓶颈是什么?
目前我国的RWE研究主要面临的是数据的问题。 具体来说包括了数据的收集和数据的整合两个方面。
对于欧美国家,电子病历、健康档案和医保数据是真实世界数据非常重要的来源。但相比于欧美发达国家,我国的电子病历系统建设起步较晚,各机构所使用的系统参差不齐,数据往往只停留在单个医疗机构内部,对数据的分析挖掘也很不足,难以发挥其真正的价值。此外,真实世界数据由于来源广泛,也导致的了数据的异质性强,数据处理难度高,不同医疗机构,不同平台间的数据很少相互连通,想要对不同来源、形式的数据进行整合面临着很大的困难。
欧洲EMA在2017年成立了关于医药大数据的特别工作小组(HMA/EMA Joint Big Data Task Force),工作组的任务包括了对于大数据的来源的确定,适用范围,有效性评估等内容,可以看出EMA对医疗大数据的重视。
前些年我们在工作中也的确曾经不时听到有些专家认为中国的真实世界数据质量不高,不能用于研究。但如果我们看一下欧美RWE研究的发展历程就不难发现,他们也一样经历了这样一个阶段,而正是通过包括政府、学术机构、企业在内的各方努力与尝试,在实践中不断的改善数据标准、数据质量和研究方法论,才有了今天真实世界数据遍地开花、各取所需的局面。如果我们因为现有数据不完整、质量有限就因噎废食,那么我们医疗大数据和RWE的工作就永远无法进步。
过去这两年,我国已经出现一些新的公司瞄准了电子病历系统,通过人工录入、图像识别、语音输入等方法,开始逐渐完善、整合各个医院的临床数据,这将为以后利用大数据进行RWE研究,起到极大的促进作用。而QuintilesIMS也立志于发挥自己在数据采集与分析领域的多年经验与特长,为中国的临床研究、真实世界研究与医疗大数据发展做出我们的贡献。






